|
CONTENIDO
INICIO
INFORMÁTICA
COMPUTADORA
HARDWARE
SOFTWARE
REDES DE COMPUTADORA
TRANSMISIÓN DE DATOS
INTERNET
BIOINFORMÁTICA
BIOLOGÍA MOLECULAR
TECNOLOGIAS DE LA INFORMACIÓN
RECURSOS DE INFORMACIÓN
BANCOS DE PUBLICACIONES.
BASES DE DATOS
HERRAMIENTAS I
HERRAMIENTAS PARA BIOLOGÍA MOLECULAR
DISEÑO DE OLIGONUCLEÓTIDOS (PRIMERS)
PCR VIRTUAL
MAPAS DE RESTRICCIÓN
ELECTROFEROGRAMAS (CHROMAS)
COMPARACION DE SECUENCIAS
DESCARGA DE PROGRAMAS DE BIOINFORMÁTICA
HERRAMIENTAS II
MODELAMIENTO DE PROTEÍNAS
APÉNDICE
OTROS CONCEPTOS
OTROS RECURSOS
PERSPECTIVAS
BIBLIOGRAFÍA
INFORMÁTICA
COMPUTADORA
Es una máquina capaz
de procesar o tratar automáticamente, a gran velocidad, cálculos y complicados procesos que requieren una toma rápida de decisiones,
mediante la aplicación sistemática de criterios preestablecidos, siguiendo las instrucciones de un programa, la información
que se le suministra es procesada para así obtener un resultado deseado.
La computadora es una maquina de propósitos o uso general. Los conceptos de estructura física y de programación constituyen el soporte material y lógico de esa realidad. Es una dualidad solidaria, que también recibe
los nombres de Hardware o soporte físico y Software o soporte lógico.
HARDWARE
Hardware son todos aquellos componentes físicos de una computadora. El
Hardware realiza las 4 actividades fundamentales: entrada, procesamiento, salida y almacenamiento secundario.
1. Entrada: Para ingresar los datos a la computadora,
se utilizan diferentes dispositivos:
-Teclado:
Dispositivo de entrada más comúnmente utilizado que encontramos en todos los equipos computacionales. El teclado se
encuentra compuesto de 3 partes: teclas de función, teclas alfanuméricas y teclas numéricas.
- Mouse: Es el segundo dispositivo
de entrada más utilizado. El mouse o ratón es arrastrado a lo largo de una superficie para maniobrar un apuntador en la pantalla
del monitor.
- Lápiz óptico: Este dispositivo es muy
parecido a una pluma ordinaria, pero conectada a un cordón eléctrico y requiere de un software especial. Haciendo que la pluma
toque el monitor el usuario puede elegir los comandos de las programas.
- Tableta digitalizadora: Es
una superficie de dibujo con un medio de señalización que funciona como un lápiz. La tableta convierte los movimientos de
este apuntador en datos digitalizados que pueden ser leídos por ciertos paquetes de cómputo. Los tamaños varían desde tamaño
carta hasta la cubierta de un escritorio.
- Entrada de voz (reconocimiento de voz): Convierten
la emisión vocal de una persona en señales digitales. La mayoría de estos programas tienen que ser "entrenados” para
reconocer los comandos que el usuario da verbalmente.
- Pantallas sensibles al tacto (Touch Screen): Permiten
dar comandos a la computadora tocando ciertas partes de la pantalla.
- Lectores de código de barras: Son
rastreadores que leen las barras verticales que conforman un código.
- Scanner: Convierten texto, fotografías
a color ó en Blanco y Negro a una forma que puede leer una computadora. También se puede ingresar información si se cuenta
con un Software especial llamado OCR (Reconocimiento óptico de caracteres).
2. Procesamiento: El CPU (Central Proccesor Unit) es el responsable de
controlar el flujo de datos (Actividades de Entrada y Salida) y de la ejecución de las instrucciones
de los programas sobre los datos. Realiza todos los cálculos (suma, resta, multiplicación, división y compara números y caracteres).
Es el "cerebro” de la computadora. Se divide en 3 componentes:
·
Unidad de control: Es en esencia la que gobierna todas las actividades de la computadora,
así como el CPU es el cerebro de la computadora, se puede decir que la UC es el núcleo del CPU. Supervisa la ejecución de los programas
Coordina y controla al sistema de cómputo, determina que instrucción se debe ejecutar y pone a disposición los datos pedidos
por la instrucción, donde se almacenan los datos y los transfiere desde las posiciones donde están almacenados. Una vez ejecutada
la instrucción la Unidad de Control debe determinar donde
pondrá el resultado para salida, ó para su uso posterior.
·
Unidad Aritmético/Lógica: Esta unidad realiza cálculos (suma, resta, multiplicación
y división) y operaciones lógicas (comparaciones). Transfiere los datos entre las posiciones de almacenamiento. Tiene un registro
muy importante conocido como: Acumulador ACC. Al realizar operaciones aritméticas y lógicas, la UAL mueve datos entre ella y el almacenamiento. Los datos usados en el procesamiento se transfieren
de su posición en el almacenamiento a la UAL. Los datos
se manipulan de acuerdo con las instrucciones del programa y regresan al almacenamiento.
·
Área de almacenamiento Primario: La memoria da al procesador almacenamiento
temporal para programas y datos. Todos los programas y datos deben transferirse a la memoria desde un dispositivo de entrada
o desde el almacenamiento secundario (disquete), antes de que los programas puedan ejecutarse o procesarse los datos.
Las
computadoras usan 2 tipos de memoria primaria:
- ROM (read only memory). Memoria de sólo lectura, en la cual se almacena ciertos
programas e información que necesita la computadora las cuales están grabadas permanentemente y no pueden ser modificadas
por el programador.
- RAM (Random access memory). Memoria de acceso aleatorio, la utiliza
el usuario mediante sus programas, y es volátil. La memoria del equipo permite almacenar datos de entrada, instrucciones de
los programas que se están ejecutando en ese momento, los datos resultados del procesamiento y los que se preparan para la
salida. Los datos proporcionados a la computadora permanecen en el almacenamiento primario hasta que se utilizan en el procesamiento.
La memoria está subdividida en celdas individuales cada una de las cuales tiene una capacidad similar para almacenar datos.
3. Almacenamiento Secundario: El
almacenamiento secundario es un medio de almacenamiento definitivo (no volátil como el de la memoria RAM). El proceso de transferencia
de datos a un equipo de cómputo se le llama procedimiento de lectura. El proceso de transferencia de datos desde la computadora
hacia el almacenamiento se denomina procedimiento de escritura. En la actualidad se pueden usar el almacenamiento Magnético
y el almacenamiento Óptico.
·
Almacenamiento Magnético: Discos Flexibles, Discos Duros y Cintas Magnéticas
o Cartuchos.
·
Almacenamiento Óptico: CD ROM (CD Read Only Memory) y WORM (Write Once, Read
Many)
·
Medios Magnético-Ópticos: Estos medios combinan las tecnologías de grabación magnética
y óptica. Un disco MO tiene la capacidad de un disco óptico, pero puede ser regrabable con la facilidad de un disco magnético.
4. Salida: Los dispositivos de salida
de una computadora es el hardware, que se encarga de mandar una respuesta hacia el exterior de la computadora, como pueden
ser:
·
Monitores: El monitor es el dispositivo de salida más común. Pueden ser que desplieguen sólo 2 colores, monitor a escala de grises y los monitores de color que pueden desplegar
de 4 hasta 1 millón de colores diferentes.
·
Impresoras: Dispositivo que convierte la salida de la computadora en imágenes impresas.
Se pueden dividir en 2 tipos: las de impacto y las de no impacto.
SOFTWARE
El Software es el conjunto de instrucciones que las computadoras emplean para manipular datos. Corresponde al
conjunto de programas, documentos, procedimientos, y rutinas asociados con la operación de un sistema de cómputo. El Software
asegura que el programa o sistema cumpla por completo con sus objetivos, opera con eficiencia, esta adecuadamente documentado,
y suficientemente sencillo de operar. El hardware por si solo no puede hacer nada, pues es necesario que exista el Software,
que es el conjunto de instrucciones que hacen funcionar al hardware.
Clasificaciones del Software
1. Sistemas Operativos: El sistema operativo (SO) es el gestor y
organizador de todas las actividades que realiza la computadora. Marca las pautas según las cuales se intercambia información
entre la memoria central y la externa, y determina las operaciones elementales que puede realizar el procesador. El SO despierta
a la computadora y hace que reconozca a la CPU, la memoria,
el teclado, el sistema de vídeo y las unidades de disco. Además, proporciona la facilidad para que los usuarios se comuniquen
con la computadora y sirve de plataforma a partir de la cual se corran programas de aplicación.
El componente más importante del SO es el Kernel, que constituye en su núcleo permitiendo la interacción entre el Hardware
y el resto del sistema; el Kernel controla los recursos del hardware, los sistemas perifericos, permite ejecutar programas
y proporciona un sistema de archivos.
+ Categorías de Sistemas Operativos.
- Multitarea. El término multitarea se refiere a la capacidad del
SO para correr mas de un programa al mismo tiempo.
- Multiusuario. Un SO multiusuario permite a mas de un solo usuario
acceder una computadora.
- Multiproceso. Las computadoras que tienen más de un CPU son llamadas
multiproceso. Un sistema operativo multiproceso coordina las operaciones de las computadoras multiprocesadores. Ya que cada
CPU en una computadora de multiproceso puede estar ejecutando una instrucción, el otro procesador queda liberado para procesar
otras instrucciones simultáneamente.
+ Sistemas Operativos más Comunes.
- MS-DOS. Es el más común y popular de todos los Sistemas
Operativos para PC. La razón de su continua popularidad se debe al aplastante volumen de software disponible y a la base instalada
de computadoras con procesador Intel.
- OS/2. OS/2 es un sistema operativo de multitarea
para un solo usuario que requiere un microprocesador Intel 286 o mejor. Además de la multitarea, la gran ventaja de la plataforma
OS/2 es que permite manejar directamente hasta 16 MB de la RAM
(en comparación con 1 MB en el caso del MS-DOS).
- UNIX-GNU/LINUX. Unix es un SO multiusuario y multitarea,
que corre en diferentes computadoras, desde supercomputadoras, Mainframes, Minicomputadoras, computadoras personales y estaciones
de trabajo. Variantes
de UNIX llevan en desarrollo o en producción más de tres décadas, haciéndolo uno de los sistemas operativos disponibles hoy
más estables, potentes, fiables y constantemente mejorado para servidores de gama alta y supercomputadoras, a la vez que sigue
siendo la solución preferida para estaciones de trabajo de alto rendimiento. UNIX fue desarrollado por Thompson y Ritchie
en AT&T Laboratories. Debido a que Unix es un Sistema Abierto, cuyo diseño interno es de dominio
Público y comercializable, existen varias versiones similares de un mismo Unix
tales como Linux, Solaris, AIX, Sinix, SCO OpenServer, SCO Unixware, Sun/OS, HP-UX, DG-UX, A-UX, Ultrix, Xenix, Centix y otros.
GNU/Linux inicio con Richard Stallman con la Free Software Foundation (1983) para desarrollar un SO de codigo
de acceso libre; a este se unio el Kernel (Linux) desarrollado por Linus Torvalds. Richard Stallman defiende la creación y
desarrollo de software libre. Este es un SO muy importante en bioinformática, sobre el cual se han diseñado la mayoría
de los programas que se utilizan en esta area.
- SISTEMA OPERATIVO DE MACINTOSH. La Macintosh es una máquina netamente gráfica. De hecho, no existe
una interfaz de línea de comando equivalente para ésta. Su estrecha integración de SO, GUI y área de trabajo la hacen la favorita
de la gente que no quiere saber nada de interfaces de línea de comando.
- WINDOWS NT DE MICROSOFT. Con Windows NT, Microsoft
ha expresado su dedicación a escribir software no sólo para PC de escritorio sino también para poderosas estaciones de trabajo
y servidores de red y bases de datos.
Microsoft Windows NT no es necesariamente un sustituto de DOS ni una nueva versión de éste; es, en conjunto,
un nuevo SO diseñado desde sus bases para las máquinas más modernas y capaces disponibles.
Windows NT de Microsoft ofrece características ínterconstruidas que ningún otro SO para PC ofrece, con excepción
de Unix. Además de las características tradicionales de estricta seguridad de sistema, red ínterconstruida, servicios de comunicación
y correo electrónico, herramientas de administración y desarrollo de sistema y una GUI, Windows NT puede correr directamente
aplicaciones de Windows de Microsoft y de Unix.
2. Lenguajes de Programación: Mediante los programas se indica a la computadora que tarea debe realizar
y cómo efectuarla, pero para ello es preciso introducir estas órdenes en un lenguaje que el sistema pueda entender. En principio,
el ordenador sólo entiende las instrucciones en código máquina, es decir, el específico de la computadora. Sin embargo, a
partir de éstos se elaboran los llamados lenguajes de alto y bajo nivel.
Los lenguajes de programación cierran el abismo entre las computadoras, que sólo trabajan con números binarios,
y los humanos, que preferimos utilizar palabras y otros sistemas de numeración.
3. Software de Uso General: El software para uso general ofrece la estructura para un gran número
de aplicaciones empresariales, científicas y personales. El software de hoja de cálculo, de diseño asistido por computadoras
(CAD), de procesamiento de texto, de manejo de Bases de Datos, pertenece a esta categoría.
+ Procesadores de Texto
Son utilizados para escribir cartas, memorandos y otros documentos, Ejemplos de procesadores de texto: Word,
AmiPro, Wordperfect.
- Hojas de Cálculo
Es una herramienta para
calcular y evaluar números. También ofrece capacidades para crear informes y presentaciones para comunicar lo que revelan
los análisis. Ejemplos de Hojas de Cálculo: Excel, Lotus 123, Quatro.
- Bases de Datos
La DBMS
(Data Base Management System) es la herramienta que las computadoras utilizan para realizar el procesamiento y almacenamiento
ordenado de los datos. Por ejemplo, un a agenda puede ser una base de datos donde se almacenan los nombres, direcciones y
números telefónicos de amigos y contactos de negocios. Ejemplos de Bases de Datos: Access, FoxPro, Approach.
- Paquetes de Presentación
Software que permite
al usuario diseñar presentaciones para desplegarlas a través de la misma computadora o imprimir diapositivas y acetatos. Ejemplos:
Presentation, Power Point, Freelance Graphics.
4. Software de aplicaciones: El software de aplicación esta diseñado y escrito para realizar tareas
específicas personales, empresariales o científicas.
REDES DE COMPUTADORA
Una Red es una manera de conectar varias computadoras entre sí, compartiendo sus recursos e información y estando
conscientes una de otra.
1. Tipos
de redes. Según el lugar y el espacio que ocupen, las redes, se pueden clasificar en dos tipos:
·
Redes LAN (Local
Area Network) o Redes de área local. Es un tipo de red que se expande en un área relativamente pequeña.
·
Redes WAN (Wide
Area Network) o Redes de área amplia. Es una red comúnmente compuesta por varias LANs interconectadas y se encuentran en una
amplia área geográfica. Entre las WAN's mas grandes se encuentran: la ARPANET,
que fue creada por la Secretaría de Defensa de
los Estados Unidos y se convirtió en lo que es actualmente la WAN
mundial: INTERNET, a la cual se conectan actualmente miles de redes universitarias, de gobierno, corporativas y de investigación.
TRANSMISIÓN DE DATOS
La transmisión de datos en las redes, puede ser por dos medios:
1. Terrestres: Son limitados y transmiten la señal por
un conductor físico.
2. Aéreos: Son "ilimitados" en cierta forma y transmiten
y reciben las señales electromagnéticas por microondas o rayo láser.
INTERNET
Existen varias
teorías relacionadas con el nacimiento de Internet, aunque la mas aceptada es que nace en los años sesenta con el nombre de ARPAnet, como un proyecto militar apoyado por el interés de las universidades que consistía en interconectar computadoras que tuvieran la capacidad de alertar
a sus ejércitos en caso de un eventual ataque. En 1972 a
las universidades de Standford, UCLA, UCSB y la de Utah se le unieron cuarenta más, dando paso a su masificación ya que se
vio beneficiada por las mini-computadoras y el UNIX, en el noventa, ya no dependía del gobierno, lo que permitió que este sistema llegara a los Hogares de la mayoría de la población.
Hoy en día Internet conecta
y ofrece servicios, tan esenciales como la propia comunicación, entre los principales encontramos el WWW o telaraña de información
mundial, el E-mail, el FTP, el CHAT y hasta el propio comercio electrónico que componen las bases de las tecnologías actuales. Una de las cosas que caracteriza a Internet es la capacidad de autosustentarse, de esta manera y orientados
al desarrollo tecnológico de las plataformas que lo componen se crean instancias de descarga de aplicaciones que favorecen
por lo general al usuario casero, ya que entregan una alternativa económica a alguna necesidad.
BIOINFORMÁTICA
Bioinformática es una
disciplina que utiliza las tecnologías de la información para captar, organizar, analizar y distribuir información biológica
con el propósito de responder preguntas complejas en biología. La bioinformática se ocupa del tratamiento de los datos en
el campo de las biociencias moleculares: biología molecular, bioquímica, medicina y biotecnología.
Según la definición del
Centro Nacional para la Información Biotecnológica National Center for Biotechnology
Information (NCBI por sus siglas en inglés): “la
Bioinformática es un campo de la ciencia en el que confluyen varias disciplinas: la biología, la computación
y las tecnologías de la información. Su fin es facilitar el descubrimiento de nuevos conocimientos y el desarrollo de perspectivas
globales a partir de las cuales puedan discernirse principios unificadores en el campo de la biología. La bioinformática,
por tanto, se ocupa de…la adquisición, almacenamiento, procesamiento, distribución, análisis e interpretación de información
biológica, mediante la aplicación de técnicas y herramientas procedentes de las matemáticas, la biología y la informática,
con el propósito de comprender el significado biológico de una gran variedad de datos. Al comienzo de la "revolución genómica",
el concepto de bioinformática se refería sólo a la creación y mantenimiento de base de datos donde se almacenaba información
biológica, como son las secuencias de nucleótidos y aminoácidos. El desarrollo de este tipo de base de datos no sólo significaba
su diseño, sino también el desarrollo de interfaces complejas donde los investigadores pudieran acceder los datos existentes
y suministrar o revisar datos. Luego toda esa información debía combinarse para formar una idea lógica de las actividades
celulares normales, de tal manera que los investigadores pudieran estudiar cómo estas actividades se veían alteradas. De ahí
surgió el campo de la bioinformática que se encarga del análisis e interpretación de varios tipos de datos, incluidas las
secuencias de nucleótidos y aminoácidos, los dominios de proteínas y su estructura.
BIOLOGÍA MOLECULAR
Los organismos presentan
una complejidad inherente que los hacen únicos, pero al mismo tiempo comparten la maquinaria básica que les da esas características
particulares. Para entender los organismos biológicos es necesario conocer sus constituyentes, las interacciones entre ellos
y el medio que los rodea. En las entidades biológicas, los bloques más básicos son las moléculas. La mayoría de estas son
moléculas inorgánicas sencillas, como sales o elementos básicos constitutivos. Otro grupo de moléculas son más complejas,
como los ácidos grasos o carbohidratos que proporcionan la energía necesaria para cumplir con las funciones vitales. Otras
macromoléculas, sin embargo, pueden diversificarse en diversas formas y funciones, desplegando una gran variedad de interacciones
y determinan las características de un organismo.
La Biología molecular es la ciencia que busca entender la forma en que los organismos trabajan y están
constituidos, desde el nivel más básico: el nivel atómico-molecular. La idea subyacente es que, para entender un organismo,
se necesita examinar sus propiedades desde el nivel más básico posible. Esta ciencia inicia con la publicación del modelo
estructural del ácido desoxirribonucleico (DNA) por Francis Crick y James Watson en 1953. Este hecho permitió conocer el proceso
en el que la información genética se transmite.
El genoma es el conjunto completo de secuencias en el material genético de un organismo. Las moléculas sobre las cuales
se centra la Biología molecular son los ácidos nucleicos,
que codifican la información genética, y las proteínas, que son las moléculas que ejecutan dicha información.
Un ácido nucleico consiste de una larga cadena de nucleótidos. La estructura básica de los ácidos nucleicos es el nucleótido.
Este tiene 3 componentes: una base nitrogenada, un azúcar y un grupo fosfato. La base nitrogenada es una purina o una pirimidina.
Estas bases se unen a la posición 1, en un azúcar pentosa, por un enlace glicosídico. Los ácidos nucleicos se denominan de
acuerdo al tipo de azúcar, el DNA tiene 2-desoxirribosa, en tanto que el ácido ribonucleico (RNA) contiene ribosa. El ácido
nucleico se construye por la unión de la posición 5´ de un anillo de pentosa, a la posición 3´ del próximo anillo de pentosa
por un grupo fosfato. Cada ácido nucleico contiene 4 tipos de bases: las purinas, adenina y guanina, están presentes en el
DNA y el RNA; las 2 pirimidinas en el DNA son citosina y timina. En el RNA se encuentra uracilo en lugar de timina. Las bases
se representan por sus iniciales. DNA contiene adenina (A), citosina (C), guanina (G), timina (T), mientras el RNA posee A,
G, C, uracilo (U). Las interacciones consisten en el establecimiento de puentes de hidrógeno, los cuales solo se pueden establecer
entre T (o U) con A (2 puentes de hidrógeno), y C con G (3 puentes de hidrógeno).
Las proteínas son cadenas de aminoácidos (compuestos orgánicos que, en los entes biológicos, contienen un grupo amino
y un grupo carboxilo) (Tabla 1) que llevan a cabo las funciones vitales de todo organismo. Estas presentan varios tipos de
estructura, la más básica es la estructura primaria, o cadena lineal de aminoácidos, determinada por la secuencia de DNA y
RNA. De acuerdo a la secuencia aminoácidica será la estructura secundaria, terciaria y cuaternaria de la proteína. Fuerzas
como puentes de hidrógeno y disulfuro, la atracción entre cargas positivas y negativas, enlaces hidrófobicos e hidrófilicos,
determinan el plegamiento que dará a lugar a hélices alfa o láminas beta, en la estructura secundaria, o intrincados modelos
en la estructura terciaria y la formación de complejos proteicos en la estructura cuaternaria.
Tabla 1. Aminoácidos y su símbolo. Están clasificados de acuerdo a su principal característica química.
|
Neutral-No polar |
Letra |
Glicina |
G |
|
L-Alanina |
A |
|
L-Valina |
V |
|
L-Isoleucina |
I |
|
L-Leucina |
L |
|
L-Fenilalanina |
F |
|
L-Prolina |
P |
|
L-Metionina |
M |
Neutral-Polar |
|
|
L-Serina |
S |
|
L-Treonina |
T |
L-Tirosina |
Y |
|
L-Triptofano |
W |
|
L-Asparagina |
N |
|
L-Glutamina |
Q |
|
L-Cisteina |
C |
|
Ácidos |
|
|
L-Aspartico |
D |
|
L-Glutámico |
E |
|
Básicos |
|
|
L-Lisina |
K |
|
L-Arginina |
R |
|
L-Histidina |
H |
La información genética
fluye por 3 procesos básicos: replicación, o copia de la información genética contenida en el DNA; trascripción, que consiste
en el traspaso de la información, desde el DNA al RNA, para que esta pueda ser finalmente llevada a proteínas, este ultimo
paso denominado traducción.
El DNA es una doble hélice,
compuesta por dos cadenas complementarias unidas entre sí por puentes de hidrogeno entre las bases nitrogenadas. La A de una hebra se aparea siempre con la
T de la hebra complementaria, y del mismo modo, la G
con la C. Durante la replicación, las dos hebras se separan
y cada una de ellas forma una nueva hebra complementaria, incorporando bases, la
A se unirá a la T de la hebra molde, la G lo hará con la C,
obteniéndose otra molécula de DNA idéntica a la original con igual información genética.
La trascripción es llevada
a cabo por una enzima, la RNA polimerasa, que reconoce secuencias
promotoras (guías moleculares) en el DNA y a partir de ellas copia una cadena de RNA (RNA mensajero o mRNA), la cual corresponde
a la región codificante (gen) para una proteína. Este proceso se denomina trascripción.
Una vez que la información
genética se encuentra como RNA es necesario llevarla a la formación de proteínas, proceso denominado traducción. Este implica
el reconocimiento del mRNA por el ribosoma, y la formación de la secuencia de aminoácidos por RNA transportadores (tRNA) a
partir de la secuencia contenida en el mRNA de acuerdo a los tripletes correspondientes en el código genético (Tabla 2).
Tabla 2. Código genético.
|
UUU |
F |
UCU |
S |
UAU |
Y |
UGU |
C |
|
UUC |
F |
UCC |
S |
UAC |
Y |
UGC |
C |
|
UUA |
L |
UCA |
S |
UAA |
Parada |
UGA |
Parada |
|
UUG |
L |
UCG |
S |
UAG |
Parada |
UGG |
W |
|
CUU |
L |
CCU |
P |
CAU |
H |
CGU |
R |
|
CUC |
L |
CCC |
P |
CAC |
H |
CGC |
R |
|
CUA |
L |
CCA |
P |
CAA |
Q |
CGA |
R |
|
CUG |
L |
CCG |
P |
CAG |
Q |
CGG |
R |
|
AUU |
I |
ACU |
T |
AAU |
N |
AGU |
S |
|
AUC |
I |
ACC |
T |
AAC |
N |
AGC |
S |
|
AUA |
I |
ACA |
T |
AAA |
K |
AGA |
R |
|
AUG |
M |
ACG |
T |
AAG |
K |
AGG |
R |
|
GUU |
V |
GCU |
A |
GAU |
D |
GGU |
G |
|
GUC |
V |
GCC |
A |
GAC |
D |
GGC |
G |
|
GUA |
V |
GCA |
A |
GAA |
E |
GGA |
G |
|
GUG |
V* |
GCG |
A |
GAG |
E |
GGG |
G |
* GUG también puede codificar para M. Este triplete es "ambiguo".
La
Biología molecular
aplica una gran variedad de técnicas moleculares, como la secuenciación de ácidos nucleicos y proteínas, la difracción de
rayos X, Reacción en cadena de la polimerasa (PCR) y clonación entre otras, lo que ha llevado a la identificación y caracterízación
de los componentes de la información genética tales como regiones promotoras, genes, operones, intrones, reguladores o dominios
de proteínas. Por ello, al estudiar todos los procesos que implican los procesos biológicos, se ha generado una gran cantidad
de información biológica que es necesario interpretar, correlacionar y difundir.
TECNOLOGÍAS
DE LA INFORMACIÓN
Las moléculas de la vida están conformadas
por una serie de componentes esenciales que, cuando se juntan, generan las propiedades específicas de las más complejas estructuras
biológicas. Estos componentes esenciales (nucleótidos o aminoácidos), son letras de un alfabeto muy especial. Estas letras
(residuos) producen patrones los cuales reflejan las propiedades de las moléculas que ellos componen, como los tripletes que
codifican para un aminoácido por ejemplo.
Resulta evidente la funcionalidad de un computador para el análisis de datos. Es así como se deduce que las tecnologías
de la información se pueden aplicar al análisis de las secuencias biológicas, como están codificadas y como se transmite entre
entidades. Las tecnologías de la información aplicadas a la biología molecular ayudan a entender como los organismos manejan
la información biológica. Esto se puede lograr determinando los patrones existentes para determinados perfiles biológicos,
y la identificación de sus similares en las diversas especies. Este proceso implica una gran atención, detalle y eficiencia.
Así, automatizando este proceso se puede generar un gran caudal de conocimiento, y determinar la información más relevante.
El amplio crecimiento de la biología molecular ha dado como resultado un auge de conocimiento, que sobrepasa la capacidad
de cualquier ser humano. Pero, si se unen los métodos computarizados al raciocinio humano, se logrará conseguir una manera
eficiente y rápida de analizar los datos existentes para extraer los detalles más significativos. Por ello, las tecnologías
de la información, unidas a la biología molecular, se convierten hoy por hoy en una de las principales herramientas para el
trabajo del científico. Este el corazón de la bioinformática.
* GUG también puede codificar para M. Este
triplete es "ambiguo".
La Biología molecular aplica una gran variedad de técnicas
moleculares, como la secuenciación de ácidos nucleicos y proteínas, la difracción de rayos X, Reacción en cadena de la polimerasa
(PCR) y clonación entre otras, lo que ha llevado a la identificación y caracterízación de los componentes de la información
genética tales como regiones promotoras, genes, operones, intrones, reguladores o dominios de proteínas. Por ello, al estudiar
todos los procesos que implican los procesos biológicos, se ha generado una gran cantidad de información biológica que es
necesario interpretar, correlacionar y difundir.
TECNOLOGÍAS DE LA INFORMACIÓN
Las moléculas de la vida
están conformadas por una serie de componentes esenciales que, cuando se juntan, generan las propiedades específicas de las
más complejas estructuras biológicas. Estos componentes esenciales (nucleótidos o aminoácidos), son letras de un alfabeto
muy especial. Estas letras (residuos) producen patrones los cuales reflejan las propiedades de las moléculas que ellos componen,
como los tripletes que codifican para un aminoácido por ejemplo.
Resulta evidente la funcionalidad de un computador para el análisis de datos. Es así como se deduce que las tecnologías
de la información se pueden aplicar al análisis de las secuencias biológicas, como están codificadas y como se transmite entre
entidades. Las tecnologías de la información aplicadas a la biología molecular ayudan a entender como los organismos manejan
la información biológica. Esto se puede lograr determinando los patrones existentes para determinados perfiles biológicos,
y la identificación de sus similares en las diversas especies. Este proceso implica una gran atención, detalle y eficiencia.
Así, automatizando este proceso se puede generar un gran caudal de conocimiento, y determinar la información más relevante.
El amplio crecimiento de la biología molecular ha dado como resultado un auge de conocimiento, que sobrepasa la capacidad
de cualquier ser humano. Pero, si se unen los métodos computarizados al raciocinio humano, se logrará conseguir una manera
eficiente y rápida de analizar los datos existentes para extraer los detalles más significativos. Por ello, las tecnologías
de la información, unidas a la biología molecular, se convierten hoy por hoy en una de las principales herramientas para el
trabajo del científico. Este el corazón de la bioinformática.
RECURSOS DE INFORMACIÓN
BANCOS DE PUBLICACIONES.
Los grandes avances en
el campo de la genómica, la proteómica y la biotecnología han catapultado a la bioinformática como una herramienta de análisis
de los grandes proyectos de secuencias, y de la innumerable cantidad de datos biológicos que se están generando. Por ello
es necesario documentarse acerca de las diferentes investigaciones y avances por medio de las publicaciones científicas que
se encuentran en Internet.
Las diferentes revistas
científicas se han agrupado en varios bancos que facilitan encontrar la información que cada investigador necesite, y en ellos
se encuentran clasificadas por temática, tipo de revista, o autor entre otros, lo que unido a poderosos buscadores facilita
enormemente su manejo. Algunos permiten acceder a sus publicaciones después de 6 meses sin restricción, pero otros solicitan
un pago por artículo (en promedio 30 dólares), generalmente con cargo a tarjeta de crédito. También hay la opción de la afiliación
por un periodo de tiempo determinado en el cuál se pueden acceder a todos las revistas del banco en particular. Asimismo,
existen en Internet editoriales, por medio de las cuales se pueden adquirir textos específicos del tema de nuestro interés
como Amazon.
1. PUBMED (www.pubmed.org): Este es el banco de publicaciones del NCBI. La página tiene, en la parte superior, una barra de búsqueda marcada
por la palabra SEARCH, donde se coloca el tema a investigar, luego de lo cual se da clic en GO, con lo que se
cargara una pagina presentando los artículos que poseen la información que se busca (Figura 1). Los artículos marcados con
un logo, que consiste en varias paginas de colores, son de acceso libre; los que tienen un logo de una sola pagina con líneas
indica que solo el resumen esta disponible (si se desea el articulo completo se debe ingresar a la pagina de la revista y
hacer el pago correspondiente); los artículos marcados con el logo de una pagina en blanco no están disponibles (ni siquiera
el resumen) (Figura 1).
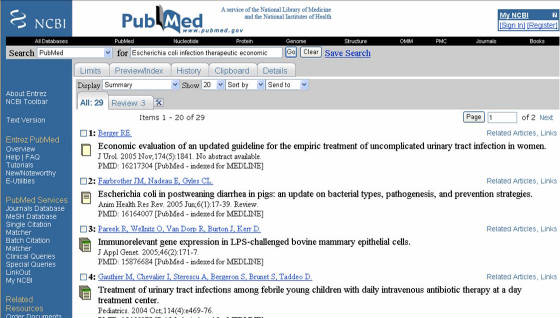
Figura 1. Pagina de resultados de publicaciones obtenidos en Pubmed. El logo de paginas de colores es para artículos completos de acceso libre,
el logo de la pagina con líneas indica libre acceso al resumen, el logo de una pagina en blanco representa artículos no disponibles.
Cuando una publicación
es de acceso libre se accede dando clic en el nombre de los autores, o en el logo (paginas de colores), luego de lo cuál se
cargará una pagina con el resumen y un link para acceder a la revista, o a la base de datos de Pubmed (Figura 2). Al hacer
clic en cualquiera de las 2 opciones saldrá una ventana emergente, donde se encuentra el artículo en formato html (Figura
3), y un link con la opción para acceder al artículo en formato pdf que se carga automáticamente.
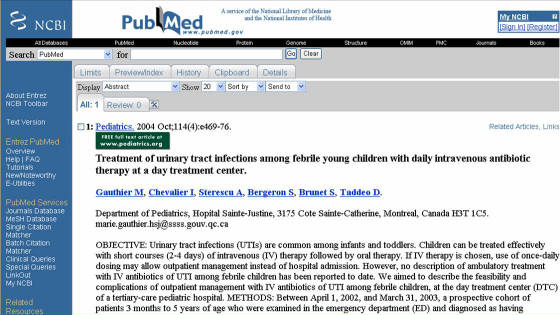
Figura 2. Pagina de Pubmed para un artículo de acceso libre. El icono verde con la leyenda “FREE full text…” en la izquierda indica
que la publicación se encuentra disponible al publico libre de pago.
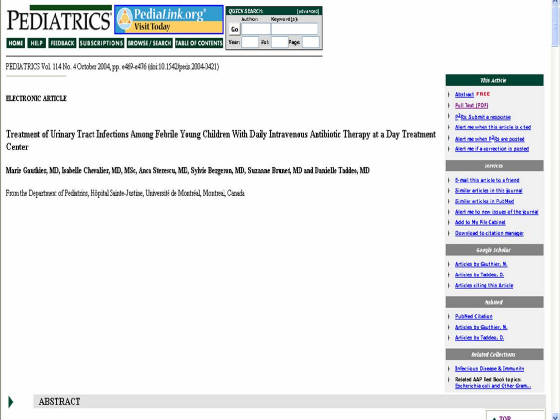
Figura 3. Pagina HTML de artículo completo. El link “full text (PDF)” se utiliza para acceder al mismo articulo en formato pdf (el segundo link en
el menú de la derecha, de color violeta).
2. ELSEVIER (www.elsevier.com): es un banco de publicaciones científicas muy amplio, que abarca recursos de gran variedad de temas (Figura
4). Contiene links a ítems como catálogos, libros, revistas, bibliografía especializada y noticias científicas destacadas,
entre otros. Un link importante se encuentra en la parte derecha, el cual enlaza al sitio Science Direct, que se puede acceder
directamente en la dirección www.sciencedirect.com (Figura 4).
Figura 4. Pagina principal de Elsevier. Contiene links a varios recursos informativos. A la derecha se encuentra el icono de acceso a Science Direct.
Por medio del sitio de Science Direct se accede a revistas y artículos
científicos (Figura 5). La página tiene una barra para la búsqueda, por tema,
revista o autor. Además, presenta todas las revistas clasificadas alfabéticamente. Es necesario pagar por los artículos o
por la suscripción, aunque hay algunas pocas revistas de acceso libre (marcados con un cuadro verde o amarillo).
Figura 5. Pagina de Science Direct. Contiene una barra de búsqueda (Quick Search), y variados links que dirige a las revistas (journals), libros (books),
resúmenes (abstracts), perfil personal (My profile) y alertas (alerts).
3. Sociedad Americana de Microbiología (ASM) (www.asm.org): Este sitio Web
presenta recursos como el calendario de eventos en microbiología, noticias y acceso a revistas científicas (Figura 6).
Figura 6. Pagina principal de la Sociedad
Americana de Microbiología. El sitio contiene enlaces a variados recursos. El motor de busqueda se inicia con el link Search en la parte superior
a la derecha.
En link SEARCH (arriba a
la derecha) abre una página con varios enlaces a revistas, junto con un formulario de búsqueda (con opciones como autor o
palabras clave) (Figura 7). La opción ASM JOURNALS busca el tema en todas las
revistas del formulario. Para iniciar el motor de busqueda se hace clic en el cuadro SEARCH.
Figura 7. Formulario de busqueda de ASM. Contiene enlaces para revistas científicas.
Si se desea acceder a mas opciones
de revistas, se da clic en SEARCH MORE
JOURNALS y en la ventana que se abre se escoge la revista de interés (Figura 8).
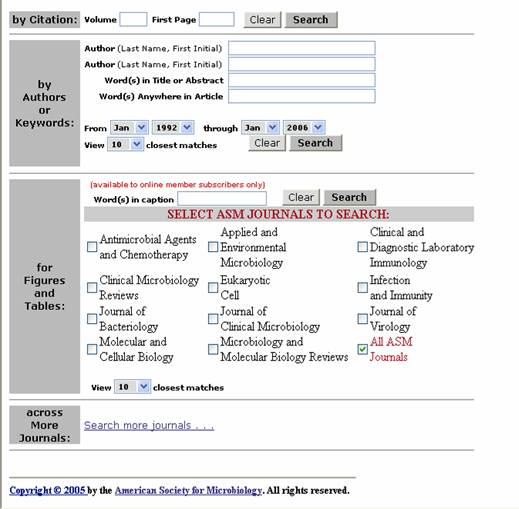
Figura 8. Formulario de busqueda de ASM. Abajo en azul esta el icono “Search more journals…” para acceder a mas opciones de revistas.
Para guardar los textos en el computador es aconsejable hacerlo en formato
pdf (Figura 9), que permite la visualización de la publicación igual a como aparece en la revista impresa, para lo cuál es
necesario bajar el programa Adobe www.adobe.com).
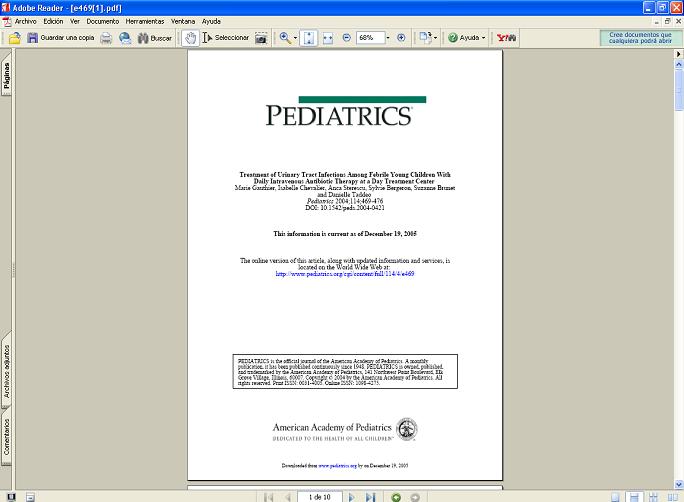
Figura
9. Articulo científico en formato pdf. La publicación se visualiza igual al impreso.
BASES DE DATOS
El gran avance de las
ciencias biologicas han generado una gran cantidad de datos, los cuales se necesita que estén disponibles para todos
los investigadores, afín de que se pueda lograr enriquecer el conocimiento, analizar la nueva información y poder establecer
correlaciones fidedignas. Por ello, desde la década de los 80 se inició la construcción de modernas bases de datos, donde
los investigadores podían enviar sus descubrimientos y además tener acceso a los logrados por otros. Inicialmente, estas solo
se ocupaban del almacenamiento de la información, pero con el tiempo se han convertido en sitios que, aparte de lograr una
eficiente organización de los datos biológicos, tienen una gran cantidad de recursos que ayudan al estudio de la información
obtenida experimentalmente. Las bases de datos son sitios de almacenamiento de información biológica de acceso libre. Las
bases de datos se han constituido en la base de las herramientas bioinformáticas. Las bases de datos son estructuradas e indexadas
lo que permite la fácil búsqueda en ellas; se actualizan periodicamente; poseen referencias cruzadas (hipervinculos) con otras
bases de datos; el almacenamiento de los datos se hace por texto plano o tablas vinculadas (flat file and relational data
base). Las bases de datos pueden ser primarias si contienen netamente datos experimentales, o secundarias si poseen datos
obtenidos a partir de la base de datos primaria.
1. Centro Nacional para la
Información Biotecnológica-NCBI (http://www.ncbi.nlm.nih.gov/): Establecido en 1988 como un recurso para la información en biología molecular,
el sitio NCBI ha creado bases de datos públicas, dirige investigación en biología computacional, desarrolla software para
análisis de datos de genomas, y disemina información biomédica. Todo esto para el mejor
entendimiento de los procesos moleculares que afectan la salud humana y la enfermedad. La ventana principal posee un listado
de links (izquierda) que dan acceso a sus diferentes subsecciones, las cuales poseen sus propias características, con la ventaja
de una interrelación entre ellas (Figura 10).
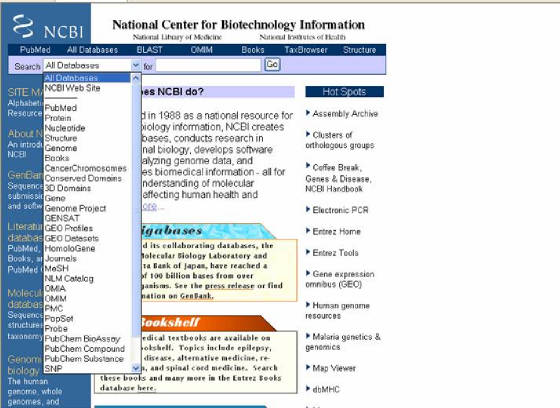
Figura 10. Pagina principal de NCBI. En la parte izquierda se ubica un menú hacia
sus principales sitios (azul), el menú que esta desplegado da acceso a diferentes recursos del sitio, el menú de la derecha
dirige hacia herramientas recomendadas.
Entre otros recursos,
NCBI tiene varias bases de datos como GenBank, OMIM (herencia mendeliana del hombre), MMDB (modelos por homología de estructuras
tridimensionales de proteínas), UniGene (Colección de secuencias de genes humanos), el mapa del genoma humano, el navegador
de taxonomía, y el CGAP (proyecto del genoma del cáncer).
Este sitio Web posee otros
recursos entre los que se cuenta el sistema de acceso integrado a secuencias, mapas, taxonomía y datos estructurales, denominado
Entrez. La literatura esta disponible a través de Pubmed. Posee el programa BLAST, el cual busca similaridades entre secuencias y es capaz de identificar
genes y sus características. También están disponibles software para la identificación de marcos abiertos de lectura (ORF),
PCR electrónica y envío de secuencias (Sequin y BankIt). Aquí se describe algunos recursos de NCBI.
- NCBI tiene el banco de secuencias biológicas más grande del mundo denominado
GenBank (Figura 11), al cual se accede haciendo clic en el icono GENBANK en el
menú izquierdo (azul) de la pagina principal. Este banco esta en colaboración con el Proyecto Internacional de Colaboración
de Base de Datos de Secuencias de Nucleótidos que se lleva a cabo en el Laboratorio
Europeo de Biología Molecular (EMBL), y con el Banco de Datos de DNA del Japón (DDBJ). Con el EMBL y el DDBJ intercambia
información diariamente para conseguir que las 3 bases de datos tengan la misma información. El sitio tiene una barra de búsqueda
en la parte superior, con un primer menú ubicado al lado de SEARCH, donde escogemos
el sitio de NCBI donde se desea que se realice el procedimiento, el cual nos da la opción de ubicar secuencias de proteínas,
nucleótidos, estructuras o en Entrez (busqueda combinada), entre otros; luego se procede a colocar el criterio de busqueda
(por medio de palabras clave, o con numero de accesión si se conoce). Por medio de GenBank se puede enviar secuencias a la
base de datos de NCBI a traves de Bankit (para presentar datos de secuencias de manera rápida) y Sequin (útil para presentaciones
complejas y extensas).
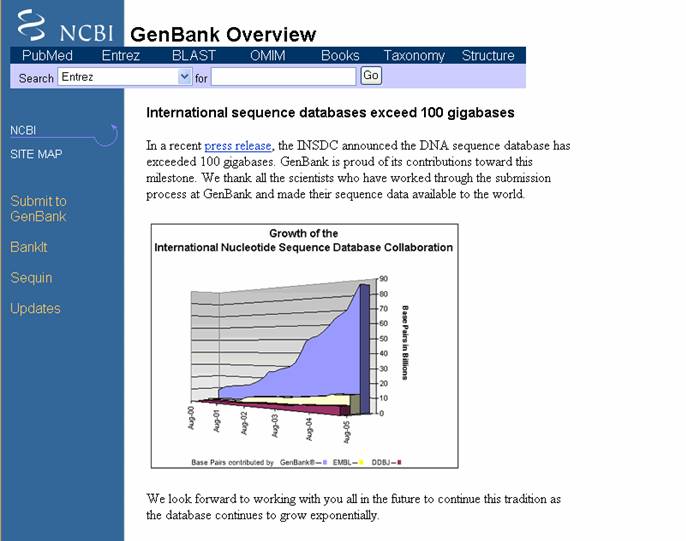
Figura 11. Pagina de GenBank.
En el listado de la izquierda (azul) se encuentran enlaces que dan acceso sitios especializados para el envío de secuencias.
- NCBI tiene la base de
datos OMIM (herencia genética mendeliana del hombre) que es un catalogo de genes y desordenes genéticos (Figura 12). Este
recurso es editado frecuentemente por el Dr. Víctor A. McKusick y sus colaboradores en Johns Hopkins y otros. Esta base de
datos contiene textos informativos sobre variados temas tales como genes relacionados con diversas enfermedades (hipertensión,
cáncer, fibrosis quística, etc.), sus localizaciones o relaciones con otros genes, entre otros. Posee enlaces para referencias
bibliograficas, mapas, secuencias y bases de datos relacionadas. Este sitio esta basado en el libro “Mendelian Inheritance in Man”. El link HELP y FAQ (en el listado de la izquierda) envía a sitios que tienen información adicional.
Figura 12. Pagina de la base de datos OMIM. Los links “Help” y “FAQ” (listado de la izquierda) proveen información detallada acerca de
la manera de usar este recurso.
- NCBI tiene la base de datos MMDB (base de datos de modelamiento molecular), que incluye biomoléculas
a las cuales se les ha determinado su modelo tridimensional por medio de cristalografía de rayos X, o por espectroscopia de
resonancia magnética nuclear (NMR) (Figura 13). Este tipo de modelos genera una gran cantidad de información acerca de la
función biológica de la molécula, mecanismos a través de los cuales lleva a cabo su función, su historia evolutiva y la interrelación
con otras moléculas. Estas estructuras provienen del Banco de Estructuras de Proteínas (PDB). Se accede a esta base de datos
por medio del link STRUCTURE, ubicado en el listado superior horizontal en el
extremo derecho, con lo cual se abre una nueva pagina que contiene un menú a la izquierda, cuyo primer link es MMDB, que da
acceso a esta base de datos. En MMDB se excluyen modelos teóricos. Esta página tiene el icono Cn3D, en el listado de la izquierda, que conduce al visor de estructuras de proteínas Cn3D (Figura 14), un programa
que permite visualizar estructuras de proteínas.

Figura 13. Pagina de MMDB. En el menú de la izquierda existen
varios recursos relacionados entre los que se destaca el programa Cn3D (abajo) para visualización de estructuras.
Figura 14. Pagina del programa Cn3D. Aquí se encuentran descritas las bondades del software y los links para su descarga e instalación.
- Otro recurso interesante es UniGene (Colección de secuencias de genes
humanos), que consiste en un sistema que busca la identificación de clusters tipo gen, en las secuencias de GenBank (Figura
15). Cada cluster contiene secuencias que representan un único gen, y además presenta información relacionada tal como tipo
de tejido donde el gen se ha expresado, y mapas de localización. Aquí se encuentran genes bien caracterizados, y muchas secuencias
que han sido expresadas recientemente. Se puede ingresar a la pagina por medio del link ALL DATABASES ubicado en la
parte izquierda del menú horizontal superior, después de lo cual se abre la pagina de Entrez, donde se escoge el enlace UniGene
(esta de cuarto en el listado derecho).
Figura 15. Pagina de UniGene.
- Existe la sección de Recursos del Genoma Humano
(Human Genome Resources), donde se entrelazan todas las secciones que contienen información relacionada con el proyecto de
secuenciación del genoma humano. Aquí hay vínculos a otras bases de datos como OMIM, UniGene o dbSNP (base de datos de polimorfismos
de un solo nucleótido). También existen links hacia mapas, citogenética y genómica comparativa. Se accede a través del enlace
HUMAN GENOME RESOURCES del listado de la derecha de la página principal de NCBI.
En esta sección se encuentra el manual de NCBI, una guía para el uso de sus recursos (menú de la izquierda) (Figura 16).
Figura 16. Pagina principal de Recursos del Genoma Humano.
- El sitio Map Viewer comprende un conjunto de
mapas, físicos y genómicos, interactivos de todos los organismos cuyo genoma se encuentra secuenciado en su totalidad (Figura
17). Map viewer despliega mapas cromosómicos, y tiene la capacidad de enfocarse en varios niveles con gran detalle, lo que
permite acceder a los datos de una secuencia en particular para una región y cromosoma de interés. Para iniciar se ubica el link MAP VIEWER en el listado derecho de la pagina y se da clic allí, con esto se abrirá otra pagina donde se encuentra el listado
de los genomas de todos los grupos de organismos que ya están terminados. Acto seguido, se escoge uno de los genomas (según
el interés particular), después de lo cual se abrirá una página que contiene un mapa cromosómico. Al escoger uno de los cromosomas
se da paso a otra ventana que contiene un mapa detallado de este, y los links para los genes identificados en él, que al seleccionarse
permiten ver la información relacionada con dichos genes (Figura 18).
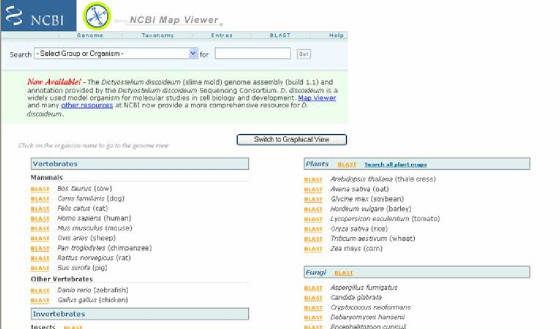
Figura 17. Sitio de Map viewer. Aparecen los enlaces hacia los genomas clasificados de acuerdo al grupo de organismo al que pertenecen.

Figura 18. Cromosoma de Map viewer. Contiene enlaces hacia páginas con información de los genes contenidos en él.
- NCBI contiene una sección sobre taxonomía,
donde se encuentran los datos que proveen información acerca de las relaciones entre organismos (Figura 19). Este sitio se
renueva constantemente según aparezcan nuevos descubrimientos que cambien los esquemas en sistemática. Contiene los nombres
de los organismos reportados en las bases de datos con al menos una secuencia (nucleótidos o proteínas). Se puede ingresar
al sitio seleccionando el enlace MOLECULAR DATABASES, y allí eligiendo el link
TAXONOMY que pertenece a Taxonomy Databases en la lista que se despliega en la
página.
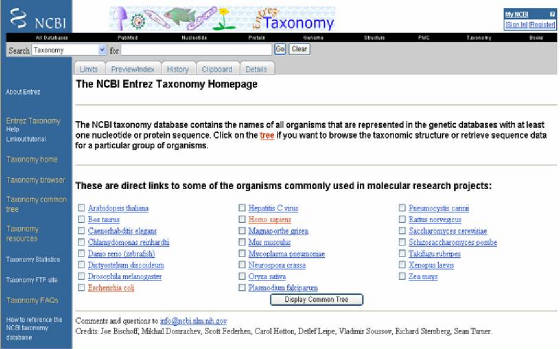
Figura
19. Ventana del sitio Taxonomy de NCBI. Aparecen los nombres de los organismos de la base de datos, que al seleccionarlos y elegir “Display Common Tree”,
se generará un árbol que representa sus relaciones.
- El proyecto CGAP (Proyecto de la
Anatomía del Genoma del Cáncer) implica la generación de información y herramientas que permitan entender
la anatomía molecular de la célula cancerigena. Este es un proyecto cooperativo entre el Instituto Nacional de Cáncer de los
Estados Unidos (NCI) y NCBI (Figura 20).

Figura 20. Pagina del Proyecto de la Anatomía del Genoma del Cáncer.
- NCBI tiene un sistema de acceso integrado
a secuencias, mapas, taxonomía y datos estructurales, denominado Entrez. Cuando se hace una búsqueda a través de Entrez, este
integra la literatura científica, bases de datos de secuencias de DNA y proteínas, estructura 3D de proteínas y datos de sus dominios, datos de estudios poblacionales, datos de expresión, montaje de genomas
completos e información taxonómica. Todo en un sistema, donde uno y otro ítem se ligan meticulosamente. Se ingresa por medio
del enlace ENTREZ HOME ubicado en el listado derecho de la página principal de NCBI (Figura 21).
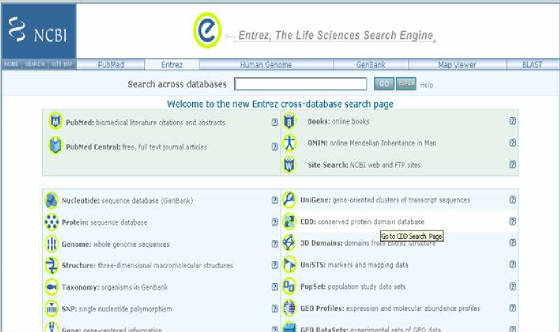
Figura
21. Sitio de Entrez. Se observa un listado que da acceso a los recursos de la base de datos de NCBI.
- Entrez es muy sencillo de manipular,
básicamente es un tutor de busqueda. Por ejemplo si escogemos el link NUCLEOTIDE tendremos
acceso a una pagina donde, si se conoce el número de accesión de una secuencia en particular (ejemplo: DQ092482) y hacemos
clic en GO, se abrirá una ventana que presentará la información acerca
de dicha secuencia y links relacionados (números de acceso, autores, revistas donde se encuentra publicado el hallazgo, titulo
del articulo, institución responsable y la secuencia) (Figura 22).
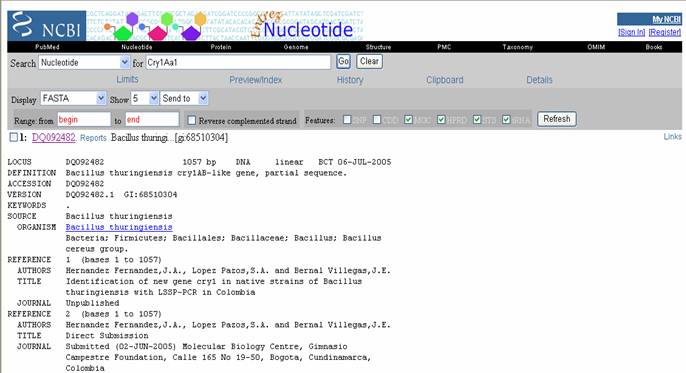
Figura 22. Resultado emitido por Entrez. Se presenta los detalles de una secuencia.
- NCBI tiene una versión
muy completa del programa BLAST (herramienta para búsqueda de alineamiento local básico), el cual busca regiones de similaridad
entre secuencias, ya sea de nucleótidos o proteínas. El programa es capaz de identificar genes y sus características gracias
a su capacidad de identificar grupos relacionados. El proceso consiste en escoger el programa según la secuencia problema
(blastn para nucleótidos, blastp para proteínas), y este comparara dicha secuencia con todas las que se encuentren en la base
de datos, indicando cuales son las más similares. El programa ayuda a identificar estructura, función, historia evolutiva
y homología de la secuencia en relación a otras, basado en estimativos estadísticamente significativos.
Se accede a través de la página principal de NCBI, haciendo clic en BLAST (en la parte superior), luego de lo cual se abre una ventana, donde se elige el ítem de acuerdo al objetivo
que se persiga (búsqueda de similaridades, alineamiento, traducción, genomas, recuperación de datos o expresión génica) y
al tipo de secuencia que se desee analizar (Figura 23).
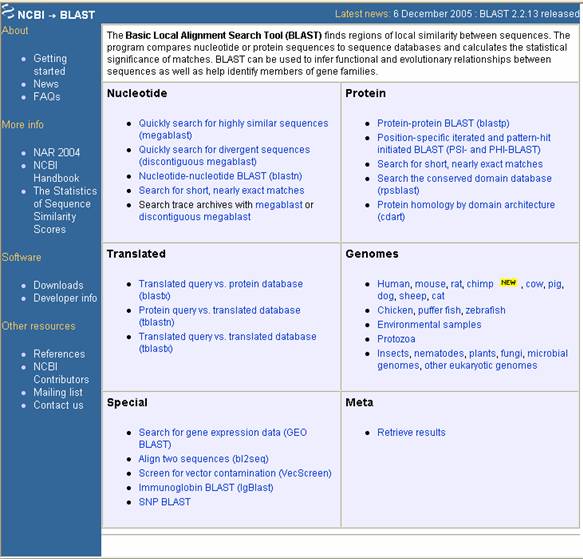
Figura
23. Ventana principal del programa BLAST. Existen varias opciones que van desde el tipo de secuencia hasta el tipo de análisis que se desee.
La principal función del programa es la búsqueda de
similaridades, con blastn o blastp. Al hacer clic en ellos se abre una ventana con un formulario donde se ingresa la secuencia
problema en la casilla SEARCH y se hace clic en BLAST (Figura 24).
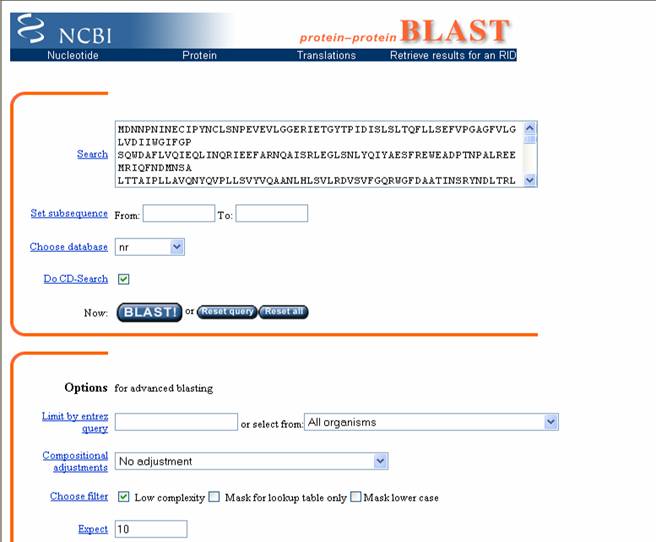
Figura 24. Formulario de ingreso de una secuencia al programa BLAST. Obsérvese que en la casilla junto a “Search” se encuentra una secuencia de aminoácidos.
La pagina que se despliega a continuación indica que la secuencia fue recibida en el servidor exitosamente,
el número de caracteres de dicha secuencia y, en el caso de una secuencia de proteína, indicará los dominios que se hayan
detectado. Después de esto se hace clic en el link azul FORMAT (Figura 25).
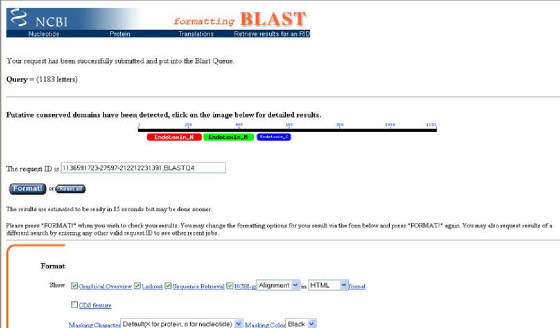
Figura 25. Pagina de resultado parcial de BLAST. Aquí se indica que la secuencia enviada fue recibida exitosamente en el servidor, y que se detectaron 3 dominios funcionales.
Luego del paso anterior, se abre una pagina de resultados donde se aprecia un gráfico que representa
todas las secuencias de la base de datos que coinciden con la que se envió, con el puntaje obtenido, representado en colores
(negro, azul, verde, púrpura y rojo de menor a mayor similitud) (Figura 26).

Figura 26. Representación gráfica de los resultados arrojados por BLAST. El nivel de identidad se representa con colores.
Debajo del gráfico se presenta un listado de las secuencias que coinciden, de mayor a menor porcentaje
de similaridad, seguido de un resumen de la denominación, número de accesión, y estimados de significancía estadística (Figura
27).

Figura 27. Listado de las secuencias con algún nivel de homología según los resultados de BLAST. Las secuencias contienen un link de acceso, una breve
denominación y los estimativos estadísticos.
Por ultimo, en la pagina se encuentran todas las secuencias que coinciden, con detalles que
incluyen su nombre y una corta definición del organismo al cual pertenece una determinada secuencia, numero de acceso en NCBI,
porcentaje de identidad, puntajes, estimados estadísticos, gaps (presentados con el símbolo “-“), y alineamiento
entre la secuencia problema con las demás (las coincidencias se indican con el símbolo “|”) (Figura 28). BLAST ayuda, por medio de esta información, a identificar una determinada secuencia, clasificarla
dentro de una familia, e inferir sus posibles relaciones evolutivas, entre otros ítem.

Figura 28. Detalles y alineamientos entre la secuencia problema y las reportadas en NCBI según el programa BLAST. Se
observa la denominación de la secuencia, el porcentaje de identidad, los gaps que se encontraron, el puntaje obtenido y el
alineamiento.
- El software para PCR electrónica es usado para identificar STS (Sequence Tagged Site: Sitio de Secuencia
Etiquetada) dentro de secuencias de DNA, mediante la búsqueda de subsecuencias con las cuales primers para PCR se alineen
correctamente (Figura 29).
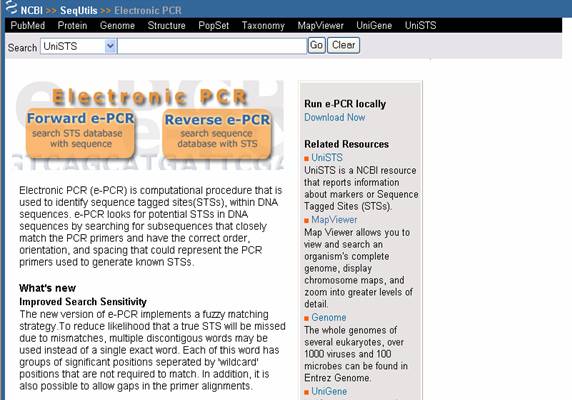
Figura 29. Pagina del software PCR Electrónica.
- En el sitio de NCBI, existe un software para
la identificación de marcos abiertos de lectura (ORF) de un tamaño mínimo en una secuencia del usuario, o de la base de datos
por medio del uso de un código genético estándar o alternativo. La secuencia de aminoácidos se puede guardar y compararse
mediante BLAST (Figura 30).
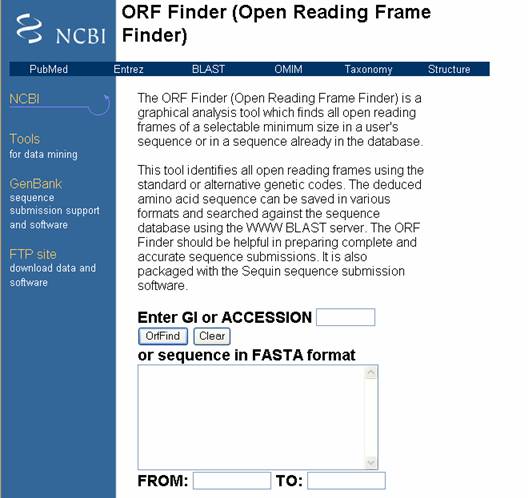
Figura 30. Pagina del software para la identificación de ORF.
- NCBI tiene otros
sitios de mucho interés como son: Genética de la Malaria, Recursos del Genoma del Ratón, o Recursos
de Retrovirus.
En resumen, NCBI es un
sitio de referencia para todo investigador. Posee múltiples sitios, con información detallada de los más importantes temas
de las biociencias. Cuenta con herramientas de primer orden, que generan una gran cantidad de información, y son fundamentales
cuando de sacar conclusiones se habla. Todo esto, unido a la interrelación entre los diferentes vínculos y bases de datos,
hace de NCBI el principal recurso Web para investigación.
2. Laboratorio Europeo de Biología Molecular-EMBL (http://www.embl.org/): El Laboratorio Europeo de Biología Molecular (EMBL) fue establecido
en 1974 y es financiado por 8 estados miembros, incluyendo casi toda Europa Oriental e Israel. Sus objetivos son dirigir investigación
básica en biología molecular, proveer servicios esenciales a científicos en sus estados miembros, dar entrenamiento de alto
nivel a su grupo investigativo, estudiantes y visitantes, además del desarrollo de nuevas herramientas para la investigación
(Figura 31).
Al
acceder a la subsección de biología computacional, en la sección de servicios, se abre una ventana con enlaces de recursos
en Bioinformática que el sitio posee. A través del Instituto Europeo de Bioinformática (EBI), EMBL abastece de servicios de
datos biológicos para la academia y la industria. Las bases de datos de EBI son:
· Banco EMBL. Fuente primaria de información de secuencias
de DNA y RNA.
· UniProt. Base de datos de secuencias de proteínas.
· Emsembl. Contiene genomas de vertebrados.
· EMSD. Base de datos de estructuras macromoleculares.
· ArrayExpress. Datos de expresión genética
basados en microarreglos.
Si se hace click en el link EMBL-EBI Services en el menu a la izquierda
de esta pantalla se accede a una ventana con links a todos los recursos del sitio (Figura 32).

Figura
31. Pagina principal de EMBL.
Aquí se presentan links que dirigen a todos los recursos del sitio.
Figura 32. Recursos bioinformáticos de EMBL.
3. Banco de Datos de DNA del Japón-DDBJ (http://www.ddbj.nig.ac.jp): El Banco
de Datos de DNA del Japón comenzó sus actividades desde 1986, en el Instituto Nacional de Genética (NIG). DDBJ es una de las
bases de datos de secuencias biológicas internacionales. Aquí se recolecta datos especialmente del Japón, aunque se aceptan
los datos de investigadores de otras orbes, y se intercambia esta información con EMBL y NCBI. También se procura el abastecimiento
de herramientas para recuperación y análisis de datos (Figura 33).
Figura 33. Ventana principal de DDBJ.
Entre las principales herramientas del sitio están:
· SRS (Sistema de Recuperación
de Secuencias). software
para búsqueda integrada de secuencias.
· TXSearch. Es un sistema para la recuperación de datos taxonómicos.
· GTOP. Contiene datos de análisis de proteínas identificadas
por varios proyectos de genomas.
· BLAST. Este sitio posee una versión del programa BLAST similar
al que provee NCBI.
· ClustalW. Esta es una herramienta de bioinformática para la búsqueda
de homología entre un grupo dado de secuencias.
· LIBRA. Es una aplicación para el análisis de secuencias y
estructuras de proteínas.
4. Banco
de Datos de Proteínas-PDB (http://www.rcsb.org/pdb/): Este sitio se encarga del mantenimiento de una base de datos de estructuras tridimensionales de proteínas, determinadas
experimentalmente por espectroscopia NMR o cristalografía de rayos x. Tiene un formulario para el envio de estructuras
por parte de los investigadores, un tutorial para aprender el manejo del sitio y acceso a los modelos por medio de códigos,
palabras clave o autor. Las estructuras contienen información completa que incluye autores, fuente, modelo de la estructura
(en varios formatos), publicación, descripción de la estructura, método experimental.
5. Recurso Universal de Proteínas (UniProt)(http://www.pir.uniprot.org/): es un catalogo de información de proteínas. Comprende información
de secuencias de proteínas y sus funciones, lo cual realiza uniendo la información contenida en Swiss-Prot, TrEMBL, y PIR. UniProt esta compuesta de 3 componentes, cada uno optimizado para diferentes
usos:
- UniProt Knowledgebase (UniProtKB) tiene extensa información curada
de proteínas que incluye función, clasificación, y referencias cruzadas.
- UniProt Reference Clusters (UniRef) database combina secuencias
cercanamente relacionadas en un record único para busquedas rápidas.
- UniProt Archive (UniParc) es un deposito que refleja la historia
de todas las secuencias de proteínas.
6. Sistema de Recuperación de Secuencias (Sequence
Retrieval System) (http://srs.ebi.ac.uk/): Este es el sitio que contiene enlaces a todas las bases de
datos. Es una base de datos que contiene bases de datos. Es el sístema idoneo para la busqueda y recuperación de secuencias
de todo tipo ya que contiene indices a todas las principales bases de datos. La busqueda de una secuencia en particular se
realiza iniciando en la sección Library Page donde se escoge la base de datos de interes, y en Query Form
se introducen las palabras claves que definen la busqueda de interes. Tiene muchos otros links de facil manejo, que con la
práctica es de mucha utilidad.
En 1988 se reunieron los staff de GenBank, EMBL y DDBJ e hicieron un acuerdo de intercambio
de información (International Collaboration of DNA sequence databases or International Nucleotide Sequence Database Collaboration-INSDC).
Así estas bases de datos intercambian información diariamente para que las tres posean los mismos datos biológicos.
PARA TENER EN CUENTA...
- Cuál es la mejor base de datos para mi proposito?
- Cuál tiene la mejor calidad de datos?
- Cuál es la más completa?
- Cuál es la más actualizada?
- Cuál es la menos redundante?
- Cuál es la más indexada?
- Cuál responde más rápido?
Y...
- Las bases de datos pueden tener muchos errores (anotaciones
automaticas)
- No todas las bases de datos están disponibles en todos
los servidores
- La frecuencia de actualización es diferente en los distintos
servidores
- La adición de datos es automatica y depende del investigador
y su veracidad
|