|
1. Modelamiento por homología de proteínas-Swiss-PdbViewer (http://www.expasy.org/spdbv/): Este software
permite el análisis de varias proteínas al mismo tiempo, ya que estas se pueden superponer para deducir alineamientos estructurales,
comparar sitios activos u otras características relevantes como mutaciones de aminoácidos, puentes de hidrogeno, ángulos y
distancias entre átomos. El programa esta ligado al servidor Swiss-Model, para modelos proteicos por homología, del Swiss
Institute of Bioinformatics-SIB, Glaxo SmithKline R&D y el Grupo de Bioinformática Estructural del Biocentro en Basel
(Figura 59).
Figura 59. Pagina principal del programa Swiss-Pdb Viewer.
Este programa es necesario descargarlo desde el sitio Web e instalarlo en el computador.
Para ello se hace clic en la opción DOWNLOAD del menú de la izquierda de la página
principal (Figura 59), con lo cual se abre una sección donde se debe aceptar un contrato de licencia haciendo clic en el enlace
I AGREE AND WANT TO DOWNLOAD SWISS PDB VIEWER NOW (en la parte inferior) (Figura
60). Luego, se abre una ventana donde tenemos la opción de elegir el sistema operativo de nuestro computador (Figura 61),
y al realizar esta acción se abre una página donde se procede a la descarga e instalación del software (Figura 62) en forma
similar a la descrita en el apartado descarga
de programas de bioinformática.
Figura
60. Pagina del acuerdo de licencia del programa Swiss-Pdb Viewer.
Figura 61. Ventana para la elección del sistema operativo del computador donde se va a instalar el programa Swiss-Pdb Viewer.
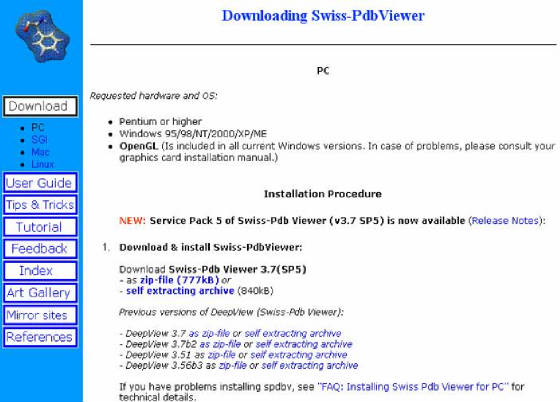
Figura 62. Pagina de descarga e instalación del software Swiss-Pdb Viewer.
Una vez que se ha instalado el programa se procede a ejecutarlo. Cuando se abre, aparece una ventana con la presentación
del software (que se procede a cerrar), seguida de otra por medio de la cual se administra las funciones de Swiss Pdb Viewer
(Figura 63).
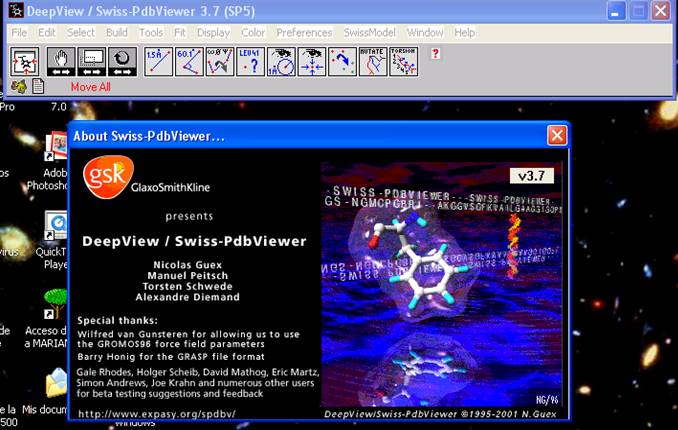
Figura 63. Ventanas de presentación y de funciones del programa Swiss Pdb Viewer. La ventana de la parte inferior tiene la presentación del programa, la
de la parte superior contiene la parte operativa del software.
Realizar un modelo 3-D
de una proteína es una tarea compleja, que incluso algunos lo han catalogado de “arte”, lo cual implica el conocimiento
profundo de la secuencia y de los objetivos que se persigan. Por ello, no se puede generalizar en reglas de diseño ya que
cada caso debe abordarse de una manera diferente. En este apartado se expone algunos pasos básicos para iniciar el modelo.
- Se guarda en una carpeta la secuencia de la proteína-problema, dentro de un archivo FASL.txt
(Figura 64), lo cual se realiza con el block de notas en el sistema operativo Windows.
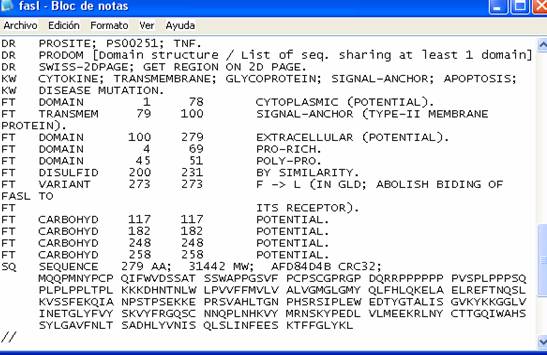
Figura 64. Secuencia de una proteína-problema en formato FASL.txt.
- A
continuación, se ingresa al software la secuencia de la proteína-problema por medio de la opción LOAD RAW SEQUENCE TO MODEL del menú SWISSMODEL, en la barra de tareas del programa. Allí se abrirá una ventana que nos permitirá acceder a la carpeta
donde este guardada la secuencia de la proteína-problema. Una vez se haya terminado esto aparecerán 2 ventanas con la secuencia,
en una como una alfa hélice, y en la otra como la estructura primaria (ventana de alineamiento) (Figura 65).
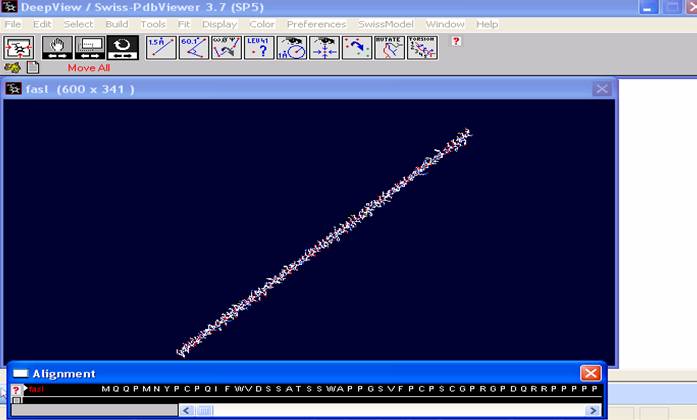
Figura 65. La secuencia de la proteína-problema una vez ingresada al programa Swiss-Pdb Viewer. La segunda ventana contiene la secuencia como una alfa-hélice,
en la ventana inferior se encuentran la secuencia de aminoácidos.
- Luego,
se elige el ítem SWISS-MODEL del menú PREFERENCES"
y en la ventana emergente se escribe el nombre del investigador y correo electrónico y se hace clic en OK.
-
Ahora se procede a escoger la opción FIND APPROPRIATE EXPDB TEMPLATES del menú SWISSMODEL, lo cual abrirá
una ventana del navegador de Internet que mostrará la secuencia de la proteína problema. Aquí se hace clic en SUBMIT. Esta acción enviará la secuencia al servidor que retornará las opciones de secuencia de proteína-molde
más adecuadas en una nueva página del navegador (Figura 66).
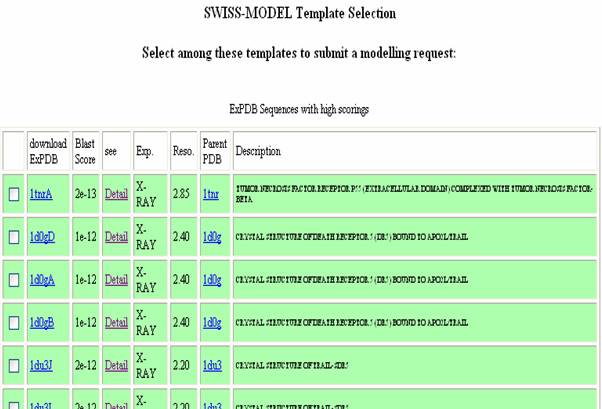
Figura 66. Lista de las posibles secuencias de proteína-molde para realizar el alineamiento con la secuencia de la
proteína-problema.
- De la lista de posibles secuencias de proteína-molde se escoge las opciones más convenientes, principalmente de acuerdo al porcentaje
de identidad, para realizar el modelamiento. Para este fin, se hace clic en el nombre de estas (Figura 66). Una vez hecho
esto se abre una ventana del computador que le solicita indicar la carpeta (Mis documentos o carpeta personal) donde se debe
guardar estas secuencias.
- Acto seguido, se ingresa las secuencias de proteína-molde elegidas al
programa por medio del menú FILE en la opción OPEN PDB FILE, lo cual abrirá una ventana del navegador que solicitara indicar la carpeta donde se guardo las
secuencias de proteína-molde. Una vez se haya hecho esto, aparecerán las estructuras 3-D al lado de la alfa-hélice de la secuencia
de proteína-problema, y en la ventana de alineamiento se podrá observar la secuencia de aminoácidos de estas (Figura 67).
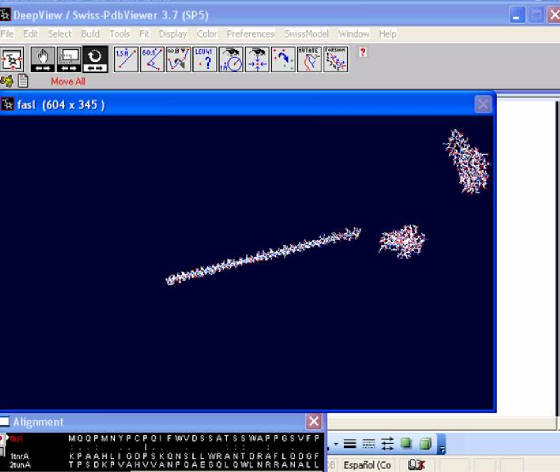
Figura 67. Secuencias de proteína-problema (alfa-hélice) y proteína-molde (3-D) una vez se han ingresado en el programa.
En la ventana de alineamiento
se encuentran las 3 secuencias de aminoácidos antecedidas por sus nombres, en rojo el nombre de la secuencia de la proteína-problema.
- En la ventana de alineamiento
se hace clic sobre el nombre de la secuencia de proteína-problema, y luego se da un clic en el marco superior de la ventana,
donde se encuentra la alfa-hélice acompañada de las estructuras 3-D de las secuencias de proteína-molde. Una vez realizada
esta acción se oprime ENTER en el teclado, con lo que la alfa-hélice desaparecerá.
- Después se hace clic
en la secuencia de proteína-molde que se desea sea utilizada como base del alineamiento (en la ventana de las estructuras
3-D deberá aparecer el nombre de esta secuencia). Acto seguido se elige el ítem MAGIC
FIT del menú FIT, se abrirá una caja de dialogo indicando que una secuencia
de proteína-molde funcionará como referencia y la otra como base, se da clic en OK.
- Se continúa con la opción
GENERATE STRUCTURAL ALIGNMENT del menú FIT,
con lo cual se realizará el alineamiento estructural (Figura 68).
- A continuación, se hace clic en el nombre de la secuencia
de proteína-problema en la ventana de alineamiento para luego elegir la opción UPDATE THREADING NOW del menú SWISS-MODEL (este ítem no es accesible si la opción UPDATE THREADING DISPLAY
AUTOMATICALLY esta activa, en cuyo caso es necesario desactivarla).
- Ahora se active la opción UPDATE THREADING DISPLAY AUTOMATICALLY
del menú SWISS-MODEL, con lo cual la secuencia de proteína-problema se alineará
alrededor de la secuencia de proteína-molde de referencia.
- Las secuencias se pueden colorear por medio del menú COLOR y el
ítem BY OTHER COLOR, previamente haciendo clic en el nombre de la secuencia que
se desee colorear en la ventana de alineamiento.
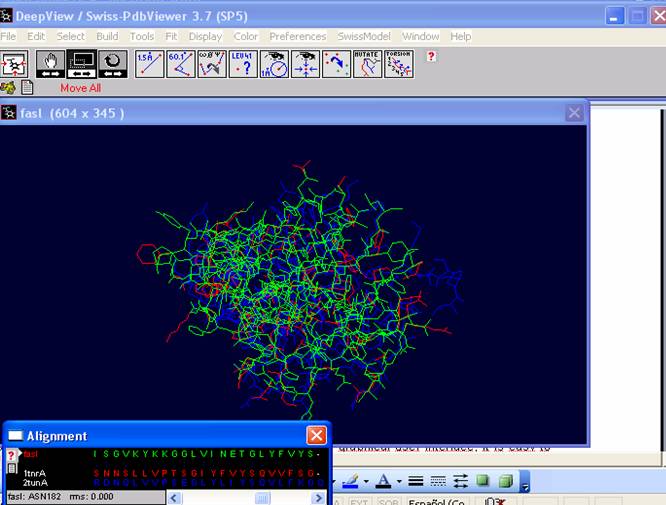
Figura 68. Alineamiento estructural de la secuencia de proteína-problema con las secuencias de proteína-molde. La secuencia de proteína-problema esta en verde, en rojo y azul se presentan las
secuencias de proteína-molde.
· Ahora se procede a enviar el modelo para su validación al servidor de Swiss Model por medio del ítem SUBMIT MODELLING REQUEST del menú SWISS-MODEL
y siguiendo las instrucciones. Por medio del correo electrónico suministrado se recibirá la respuesta (esto dura aproximadamente
de 5
a 10 minutos).
· Todo el proceso de modelamiento implica estar conectado a Internet.
El programa tiene muchas opciones para modelar, analizar, reformar y evaluar los modelos realizados, que deberán utilizarse
de acuerdo a cada caso. La página de este software tiene una guía de usuario, un tutorial básico y además de un enlace
hacia un tutorial preparado por el profesor Gale Rhodes del departamento de química de la Universidad de Southern
Maine (http://www.usm.maine.edu/~rhodes/SPVTut/index.html), el cual aconsejo estudiar minuciosamente afin de conseguir un mayor provecho de las ventajas del programa.
2. Modelamiento por homología de proteínas-Servidor Robetta (http://www.robetta.bakerlab.org): Este servidor provee
herramientas automatizadas para la predicción de estructura de proteínas y su análisis. Para la predicción de estructuras,
las secuencias son enviadas al servidor donde se arreglan en dominios y modelos estructurales usando los metodos de modelamiento
comparativo o predicción de estructuras de novo. Si se da un apareamiento confiable de la secuencia problema a una
proteína de estructura conocida por medio de BLAST, PSI-BLAST, FFAS03 o 3D-JURY, esta se usa como molde para la construcción
de un modelo por homología. Si no se da un apareamiento con ninguna estructura proteíca conocida, la predicción del modelo
se realiza usando el metodo de novo inserción del fragmento Rosetta. Asimismo, se pueden enviar datos experimentales
obtenidos de Resonancia Magnetica Nuclear (NMR), con la secuencia problema para la determinación de la estructura por RosettaNMRdenovo.
El sitio incluye la predicción de efectos de mutaciones en interacciones proteícas. Para su utilización adecuada es necesario registrarse para lo cual es necesario poseer un correo electrónico
perteneciente a una institución educativa (simbolizado por EDU en la dirección,
ejemplo xxx@universidad.edu.co).
Una vez hecho el registro se procede a ingresar al sitio por medio del enlace LOGIN
en la pagina principal (Figura 69). Luego, se puede enviar un trabajo por medio de la opción SUBMIT del menú STRUCTURE PREDICTION y se continúa llenando el formulario.
La secuencia de proteína-problema debe estar en formato Fasta. El proceso de envío de la secuencia es bastante sencillo, pero
los resultados demoran mucho (aproximadamente entre 1 y 2 meses). Robetta genera modelos 3-D confiables y los resultados contienen
amplia información. Depende de cada caso su interpretación.

Figura
69. Pagina principal del servidor Robetta.
3. El Sistema Experto en Análisis de Proteínas-Expasy (http://ca.expasy.org/): Aquí encontramos una gran variedad de herramientas para el trabajo en proteomica, principalmente al análisis
de secuencias y estructuras de proteínas y electrofóresis en gel de poliacrilamida bidimensional (2-D PAGE) (Figura 70). Este
sitio hace parte del Instituto Suizo de Bioinformática.
Contiene varias secciones:
· Bases de datos: Swiss-Prot, TrEMBL, PROSITE, SWISS-2DPAGE, ENZYME.
· Herramientas y paquetes de software: herramientas para
proteomica y análisis de secuencias, ImageMaster/Melanie.
· Educación y servicios.
· Documentación.
· Enlaces a listas de recursos de biología molecular.
· Enlaces a los grandes servidores de biología molecular.
· Misceláneos.
Figura 70. Pagina principal de Expasy.
|