|
HERRAMIENTAS I
HERRAMIENTAS PARA PCR
DISEÑO
DE OLIGONUCLEÓTIDOS (PRIMERS)
El primer (oligonucleótido o iniciador) actúa como punto
de anclaje para la DNA polimerasa, y como un iniciador de la reacción de replicación del DNA. En el laboratorio,
se utilizan para dar inicio a reacciones de replicación en puntos deseados de una muestra, generalmente para realizar reacciones
de secuenciación, o amplificar el número de copias de una muestra de DNA (PCR).
1. Características de un primer: un primer debe ser específico para la región que se desea replicar,
mantener la suficiente energía necesaria para las condiciones experimentales, y evitar la formación de estructuras que puedan
impedir la reacción.
La eficiencia de la PCR depende en gran medida de los oligonucleótidos que se diseñen. Los oligonucleótidos deben ser
diseñados cuidadosamente. Generalmente se ha utilizado el sentido común para ello. Es importante que se sigan unas reglas
mínimas de diseño que permitan su especificidad para una determinada región de DNA.
Las reglas básicas de diseño son
· Longitud. Pueden ser muy cortos o muy largos. Como
norma general se acepta que su tamaño máximo sea de 30 bases. Pero los mejores oligonucleótidos están comprendidos entre 18
y 25 pares de bases, que hacen que la temperatura de melting oscile entre 50 y 600C
(temperatura óptima para el alineamiento).
· Contenido
de G-C. Para determinar este
parámetro es necesario conocer el contenido de G-C de la secuencia a amplificar, pero como regla básica se prefiere oligonucleótidos
con un contenido de GC de 40 a 60%, que les da estabilidad
y una temperatura de melting apropiada.
· Se debe evitar que existan más de 3 repeticiones
consecutivas de una base en su diseño (ej. AAAAA).
· No deben tener purinas o pirimidinas repetidas consecutivamente más de 3 veces en la medida
de lo posible.
· La temperatura de melting debe ser lo mas
parecida posible (máximo 5 grados de diferencia entre uno y otro primer).
· Es importante observar los extremos 5’ y 3’
de los oligonucleótidos y revisar que estos extremos no sean complementarios, ejemplos:
a) 5’
NNNNNNNNNNNNNNTATA 3’
5’
NNNNNNNNNNNNNNTATA 3’ à 5’
NNNNNNNNTATA 3’
3’ATATNNNNN5’
à DIMERO
DE PRIMERS.
b) 5’ TATANNNNNNNNNNNNN 3’
5’ TATANNNNNNNNNNTATA
3’ à 5’
TATANNNNN 3’
3’ NNNNNNNNNNATAT 5’
à NO HAY EXTENSION
c) 5’ NNNNNNNNNNGCATGC 3’
à 5’ NNNNNNNNGCA 3´
3’CGT
se forma 1 horquilla
à PRODUCTOS DE PCR NO DESEADOS.
2.
Diseño de primers para
aislamiento de proteínas (Primer3) (http://frodo.wi.mit.edu/primer3/primer3_code.html): Primer3 es un programa escrito
para el diseño automatizado de primers para PCR. Primer3 compara una secuencia de DNA molde con una secuencia de DNA, o proteína,
para generar secuencias de primer directo, calculando todas los posibles oligonucleótidos específicos para una determinada
región. Finalmente, Primer3 genera las correspondientes secuencias de primer reverso, además de calcular otra información
necesaria como la temperatura de melting, o el contenido de GC de cada primer.
La página principal (Figura 34) contiene la presentación del software. Para acceder al programa se elige TRY THE WEB
INTERFASE (Figura 34).
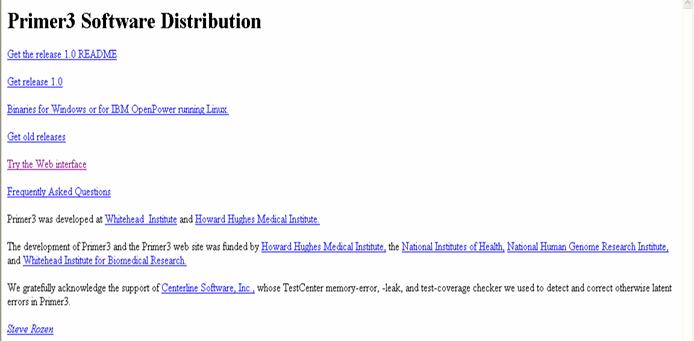
Figura 34. Pagina principal del programa Primer3. Se encuentra aquí una presentación del software, para ingresar
al programa se hace clic en “Try the Web interfase” (violeta).
La ventana principal del programa presenta varias características, que son muy útiles para un diseño adecuado del primer.
Cada ítem es un enlace, hacia una pagina de ayuda, donde se amplia la información para su utilización (Figura 35).El programa
tiene, en la parte superior, una caja donde se coloca la secuencia para la cual se desean los primers. En este lugar, existe
la opción de escoger una de las librerías disponibles (ninguna, humano, roedor y simple, roedor o drosophila), que tienen
la finalidad de evitar que el primer se diseñe sobre sitios repetitivos en la secuencia (especialmente microsatélites). Debajo
se presentan 3 cuadros. El primero y el tercero se seleccionan para el primer directo (LEFT PRIMER) y reverso
(RIGHT PRIMER). El segundo cuadro se escoge si se desea diseñar una sonda de hibridación (HYBRIDIZATION
PROBE) (Figura 35). Si se desea, con esta información
básica se puede ordenar al programa el diseño de los primers haciendo clic en PICK PRIMERS (Figura 35).
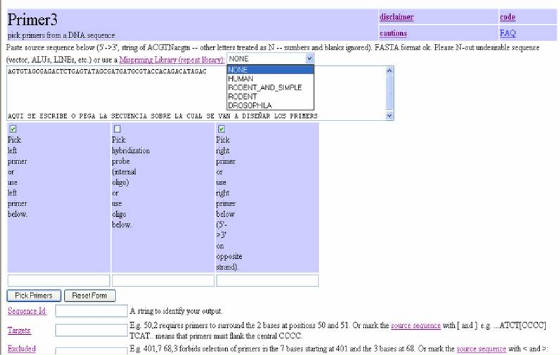
Figura 35. Programa Primer3. Se observa
la parte superior del formulario.
Una segunda parte del programa (Figura 36) provee herramientas que permiten acomodar el diseño final de los primers
a unas características específicas, que dependerán de los objetivos de la investigación. Una primera opción, en esta sección,
es la identificación de la secuencia (SEQUENCE ID) que puede hacerse con números o letras. Luego
sigue una casilla (TARGETS) donde se coloca una región en especial la cual se desea que los
primers flanqueen. En tercer lugar esta el ítem EXCLUDED REGIONS, donde se puede excluir del
diseño alguna región dentro de la secuencia. Con la casilla PRODUCT SIZE RANGES se pretende elegir el tamaño del producto que los primers van a flanquear
(se debe colocar un rango). Si lo que se busca es un tamaño muy especifico de producto se elige CLICK HERE TO SPECIFY THE MIN, OPT, AND MAX PRODUCT SIZES ONLY IF YOU ABSOLUTELY
MUST. USING THEM IS TOO SLOW
(AND TOO COMPUTATIONALLY INTENSIVE FOR OUR SERVER). NUMBER TO RETURN
se utiliza para escoger la cantidad de opciones, en pares de primers, que se quiere que el programa arroje. MAX 3'
STABILITY sirve para determinar la estabilidad de las bases del extremo 3’ de los primers. En el caso de la opción
MAX MISPRIMING, esta se puede utilizar para determinar el máximo apareamiento con una región
de las librerías. La opción PAIR MAX MISPRIMING indica el máximo permitido de suma de similaridades de un par de primers, con alguna secuencia
en las librerías. Si solo se desea definir estas características
se puede hacer clic en PICK PRIMERS (Figura 36).
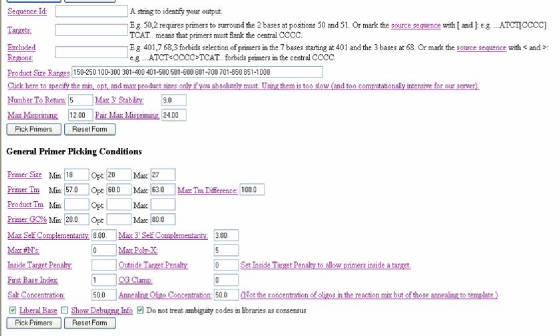
Figura 36. Programa Primer3. Se observa la segunda sección del formulario.
Una tercera sección en esta ventana se denomina GENERAL PRIMER PICKING CONDITIONS, que nos da otras opciones de diseño. La primera, PRIMER SIZE, permite elegir la longitud de
los oligonucleótidos. PRIMER TM define la temperatura de melting de los oligonucleótidos. MAX TM
DIFFERENCE se utiliza para escoger la diferencia de Temperatura de melting entre uno y otro primer (se aconseja que
esta diferencia sea la más pequeña posible). PRODUCT TM ayuda a escoger la temperatura de melting del amplificado
o producto. PRIMER GC% sirve para escoger el porcentaje de GC en los primers. El ítem MAX SELF COMPLEMENTARY
permite definir el nivel de autocomplementaridad entre cada par de primers, en tanto que MAX 3’
SELF COMPLEMENTARY define el puntaje de autocomplementaridad en el extremo 3’. En el apartado MAX #N’S se puede elegir el número máximo
de bases desconocidas (N) para un primer. En cuanto a MAX POLY-X,
referencia la máxima cantidad en repeticiones de una base para un primer (Ej. AAAAA). INSIDE TARGET PENALTI
permite al programa incluir posiciones sobrelapadas. La función del parámetro OUTSIDE TARGET PENALTI es la
de incluir regiones cercanas a la secuencia blanco. El punto FIRST BASE INDEX es para incluir la primera
base de la secuencia aportada. El objetivo del apartado GC CLAMP es definir el número de Gs y Cs consecutivas
en el extremo 3’ de la pareja de primers.
SALT CONCENTRATION es usado para calcular la temperatura de melting, en base a la concentración de sales
(usualmente KCl). El argumento ANNEALING OLIGO CONCENTRATION ayuda a que se pueda determinar la temperatura
de melting de los oligonucleótidos, en correspondencia a un protocolo estándar de PCR. En cuanto al parámetro LIBERAL
BASE ayuda a que Primer3 acepte códigos IUPAC o IUB para bases ambiguas (N). Si se han definido estas condiciones
para los primers, se puede proceder a hacer clic en PICK PRIMERS (Figura 36).
Más abajo, en la pagina del programa (Figura 37) existen otras opciones, como la de incluir una región en especifico (INCLUDED
REGION); determinar la primera base de un codón de inicio (START CODON POSITION), afín de definir
marcos de lectura; la calidad de la secuencia (QUALITY SEQUENCE) y OBJECTIVE FUNCTION PENALTY WEIGHTS
FOR PRIMERS que ayudan al usuario a variar algunos de los parámetros del
programa predeterminados. Finalmente, se encuentra la sección HYB OLIGOS (INTERNAL OLIGOS), que sirve para el diseño de oligonucleótidos internos usados para detección basada
en hibridación, cuyas características son similares a las utilizadas para el diseño de primers.
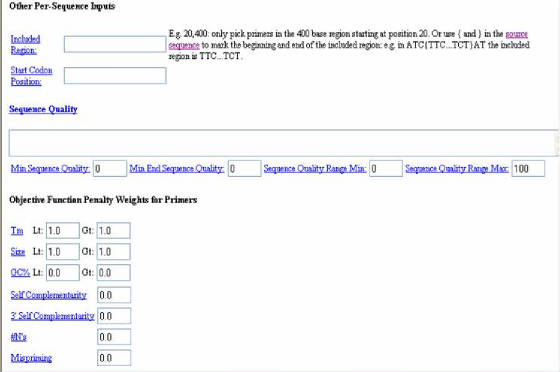
Figura
37. Programa Primer3. Se observa la tercera parte del formulario.
Cuando
hemos definido todos los parámetros necesarios del programa y se le indica que diseñe los primers, el sitio procesa la información
y arrojara una ventana con varias opciones de oligonucleótidos. Generalmente, la primera es la mejor, pero será la experiencia,
conocimiento y el sentido común del investigador lo que le llevara a escoger una de ellas.
3. Diseñando
primers para mutagénesis dirigida (PrimerX)(http://bioinformatics.org/primerx/): Primer X es un programa para automatizar el
diseño de primers de PCR mutagénica, con el fin de realizar mutagénesis sitio-dirigida. PrimerX compara una secuencia molde
de DNA con una secuencia de DNA, o proteína, a las que se les incorpora la mutación deseada. El programa genera secuencias
de primers directos computando todas las posibilidades de oligonucleótidos que posean la longitud apropiada, que codifiquen
para la mutación y genere el producto específico para el cual fue diseñado. Finalmente, PrimerX genera las secuencias de primers
reversos correspondientes, y calcula otra información necesaria, como temperatura de melting y contenido de GC para cada par
de primers (Figura 38).

Figura 38. Pagina principal del programa PrimerX.
· Diseño de primers basado en secuencia de
DNA. PrimerX diseña primers mutagénicos basado en 2 clases de secuencias de datos. Una opción consiste en ingresar una mutación
en su secuencia de DNA molde, de esta forma las inserciones, delecciones o sustituciones de bases son incorporadas. Aquí,
el programa contiene 2 maneras de ingresar la secuencia, la primera consiste en subirla desde el disco duro del computador
utilizando el ítem EXAMINAR (la secuencia debe estar en formato FASTA); la otra alternativa es pegar la secuencia
en el cuadro ubicado debajo del ítem anterior. Luego de haber realizado esta tarea, se procede a ingresar el código de la
mutación que se desea según el formato siguiente:
Mutación
Código de Mutación
Descripción
Sustitución
C15G
Reemplazar a "C" en la posición
15 con
"G".
CG15AT Reemplazar
"CG" en las
posiciones 15-16
con "AT".
Delección C15del
Quitar a "C" en la posición 15.
CG15del
Quitar "CG" en las posiciones
15-16.
Inserción C15insG
Insertar a "G" en la posición 15,
corriendo a "C" a la posición
16
CG15insAT Insertar
"AT" en las posiciones
15-16,
corriendo "CG" a las
posiciones 17-18
Después que se ha completado estas
acciones, se prosigue a escoger el protocolo de mutagénesis entre las opciones que da el programa (que van desde definido
por el usuario hasta las variantes comerciales). Cuando se ha terminado se hace clic en NEXT (Figura 39), con lo cual se abre una ventana donde el programa solicita la especificación de algunos parámetros
como la temperatura de melting, el contenido de GC, la longitud de los primers entre otros. Una vez se ha realizado esta ultima
parte se da clic en GENERATE PRIMERS (Figura 40). El programa mostrará
una ventana con los resultados.
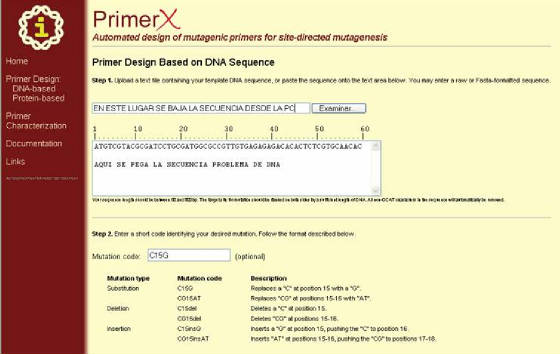
Figura 39. Pagina del programa PrimerX para el diseño de primers basado en secuencia de DNA. Una vez se ha
completado este formulario se hace clic en “Next” (al final del formulario)
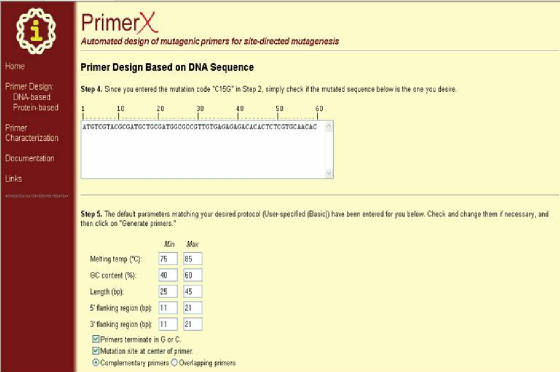
Figura 40. Segunda página del programa PrimerX para el diseño de primers basado en secuencia de DNA. En esta ventana se ingresa los parámetros necesarios y se hace clic en “Generate primers” (al final del formulario).
·
Diseño de primers basado en secuencia de proteína. Otra opción consiste en ingresar
una mutación en la secuencia de la proteína, codificada por su molde de DNA, en cuyo caso PrimerX genera primers mutagénicos
basados en todas las posibles secuencias de DNA que codifican la mutación deseada, teniendo en cuenta el código genético.
Se recomienda esto para cambiar un aminoácido específico en otro. En primer lugar se procede a ingresar la secuencia de DNA
de la manera ya descrita para, y se hace clic en TRASLATE, con lo que se abre una ventana donde se presenta
la secuencia de aminoácidos correspondiente a la secuencia de DNA que se ingreso, y se pide el código de mutación de acuerdo
al formato siguiente:
Mutación
Código
Descripción
Sustitución
E15P Reemplazar "E" en
posición
15 con "P".
EF15PQ Reemplazar "EF" en
posiciones 15-16 con
"PQ".
Delección
E15del Quitar "E" en la posición
15.
EF15del
Quitar "EF" en las
posiciones 15-16.
Inserción
E15insP
Insertar "P" en la
posición 15, corriendo la
"E" a la posición 16.
EF15insPQ
Insertar "PQ" en las
posiciones 15-16,
corriendo "EF" a las
posiciones 17-18.
Después
de lo anterior, se escoge el protocolo de mutagénesis, igual a como ya se describió y se hace clic en NEXT
(Figura 41).
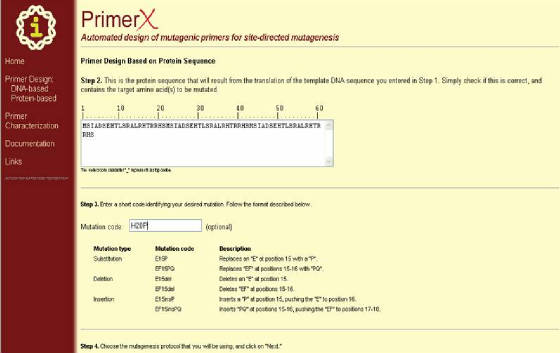
Figura 41. Pagina del programa PrimerX para el diseño de primers basado en secuencia de proteína. Se usa en forma
similar a la manera en que se utiliza con secuencia de DNA, al cabo de lo cual se hace clic en “Next” (al final
del formulario).
Una vez completado
lo anterior se abre una ventana que nos pide algunas especificaciones (según el protocolo de mutagénesis elegido) como por
ejemplo el sistema de expresión a utilizar, la temperatura de melting, el contenido de GC, o la longitud de los primers entre
otros. Una vez definido lo anterior se hace clic en GENERATE PRIMERS, con lo cual el programa arrojará los resultados (Figura 42).
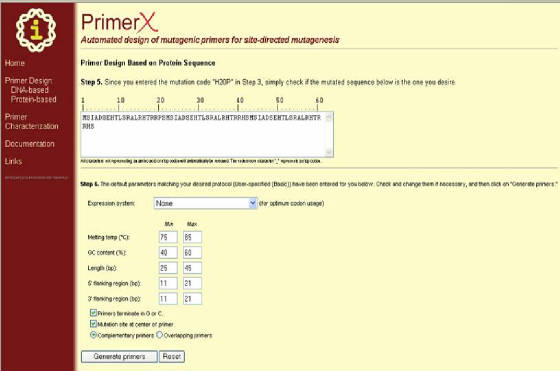
Figura 42. Segunda página del programa PrimerX para el diseño de primers basado en secuencia de proteína. En esta ventana
se ingresa los parámetros necesarios y se hace clic en “Generate primers” (al
final del formulario).
Caracterización
de secuencias de primers mutagénicos. Además de lo anterior PrimerX puede caracterizar primers que usted ha diseñado. Aquí,
se necesita únicamente ingresar una secuencia de primer mutagénico, y el número de bases que no se corresponden (Figura 43),
y PrimerX calculará y devolverá su complemento reverso, contenido de GC, o temperatura de melting (Figura 44).
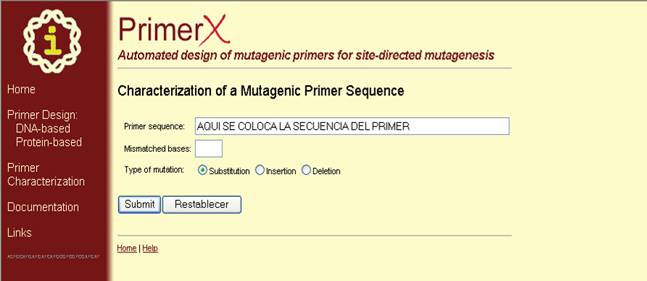
Figura 43. Caracterización de secuencias de primers mutagénicos. En esta
ventana se ingresa la secuencia del primer y las bases alteradas y se envía por medio de la opción “Submit”
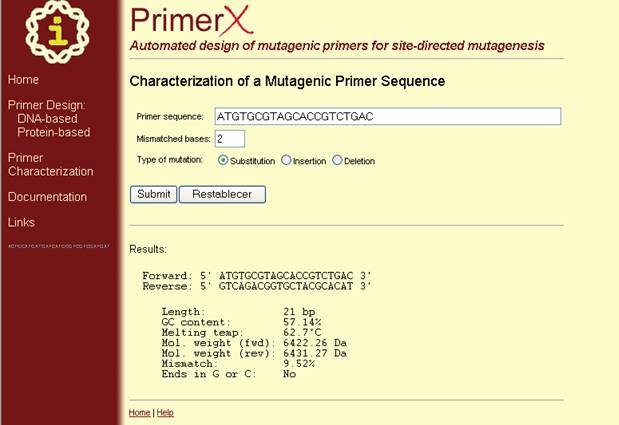
Figura 44. Pagina de resultados del programa PrimerX en caracterización de secuencias de primers mutagénicos. En esta ventana se observa las condiciones calculadas para
el primer caracterizado.
PCR-VIRTUAL(http://www.ch.embnet.org/software/iPCR_form.html): La técnica de PCR utiliza una enzima, llamada polimerasa, para multiplicar rápidamente un pequeño fragmento de DNA. Cada
ciclo de PCR consta de tres fases. En la primera, llamada denaturación, se calienta el DNA para separar las dos cadenas que
lo forman. En la segunda, llamada alineamiento, la temperatura de la mezcla se baja para que los primers u oligonucleótidos se enlacen con las cadenas separadas de esta molécula. En la tercera, o polimerización,
se eleva de nuevo la temperatura para que la enzima polimerasa copie rápidamente el DNA. En cada ciclo de PCR se duplica todo
el DNA presente en la reacción, de manera que en unas pocas horas se obtienen más de mil millones de copias de un solo fragmento.
PCR
virtual es un programa interactivo escrito para automatizar la PCR, muy útil para evaluación del diseño de primers. La página
contiene un formulario con varios ítems (Figura 45). La herramienta se maneja de la siguiente forma:
- En la primera caja (SELECT FORMAT) se debe escoger EMBLID
OR AC si tenemos el número de accesión de la secuencia en base a la cual diseñamos el primer. Si no es así, es necesario
pegar la secuencia, para lo cual se escoge el ítem PLAIN TEXT.
- En la siguiente caja, QUERY TITLE
(OPTION), se le puede asignar un nombre. Luego, continúa la caja PASTE YOUR SEQUENCE HERE,
donde se pega la secuencia de DNA, o en su defecto se escribe el número de accesión de la base de datos.
- Posteriormente, en la caja PASTE
FORWARD PRIMER se escribe la secuencia del primer directo, y en la caja
PASTE REVERSE PRIMER se copia la secuencia del primer reverso. En la sección OPTIONS están
2 cajas. En la primera, MINIMAL AMPLIFIED LENGTH, se coloca
el mínimo tamaño del fragmento que se desea amplificar, y en la siguiente MAXIMAL AMPLIFIED LENGTH,
el máximo tamaño del fragmento que se va amplificar.
- Finalmente, se hace clic en la opción
RUN PCR. Los resultados que da el programa indican la secuencia que amplifica, el tamaño del producto y los
oligonucleótidos empleados (Figura 46).
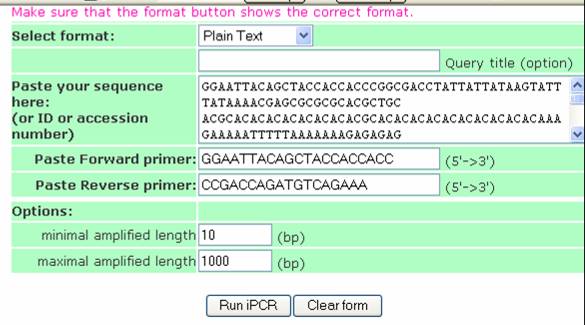
Figura 45. Pagina del programa de PCR virtual. Contiene un formulario donde se ingresa la secuencia problema y los primers diseñados para ella, además
se define el tamaño del amplificado.
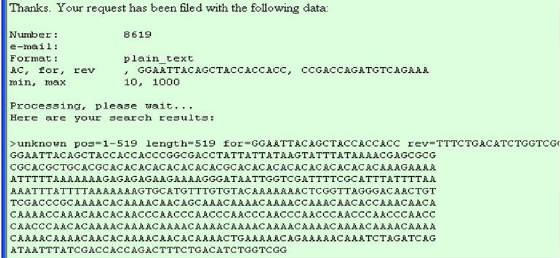
Figura
46. Pagina de resultados del programa de PCR virtual. Contiene los primers, y la secuencia que resulta de la amplificación
virtual.
MAPAS DE RESTRICCIÓN (http://arbl.cvmbs.colostate.edu/molkit/mapper/): El mapeo se refiere a la construcción de un mapa de los fragmentos
de una secuencia. La meta es predecir los fragmentos que se pueden generar cuando la secuencia ha sido cortada con un reactivo,
o conjunto de ellos.
Para estudiar mejor una macromolécula es muy útil fragmentarla en pequeñas piezas, y estudiarlas
por separado. Esto simplifica el trabajo, y evita perder información de interacciones entre las partes de la macromolécula.
Esto es muy usado para DNA y proteínas.
Estos mapas ayudan a dirigir la investigación, permitiendo acciones como mutagénesis sitio-dirigida,
o la detección de la presencia de una molécula o secuencia dada. La fragmentación de una molécula de DNA se realiza normalmente
por medio de enzimas de restricción. Estas enzimas reconocen un patrón específico de nucleótidos en la secuencia, se une a
este y corta en una posición relacionada al sitio de reconocimiento. El reconocimiento sobre la secuencia se da en un conjunto
de 4
a 6 nucleótidos y frecuentemente son reconocidos por más de una enzima. Las proteínas son fragmentadas
utilizando una variedad de métodos, entre los que se cuentan enzimas especiales (proteasas), o el tratamiento con ácidos fuertes
y una variedad de reactivos químicos.
Aquí se describe un programa interactivo para la realización de mapas de restricción de DNA
(Figura 47). El sitio consta de una sola página dividida en 2 secciones. La primera (superior) describe el programa, e instruye
en su utilización (Figura 47). Las enzimas a utilizar se pueden elegir de acuerdo a:
· Todas
las enzimas de restricción:
enzimas en la base de datos.
· Enzimas
más frecuentes: enzimas comúnmente
utilizadas para múltiples sitios de clonación, o las que el autor ha favorecido.
· Cortes
romos: enzimas que clivan para
generar extremos romos.
· Cortes
únicos: enzimas que cortan una
sola vez en el DNA blanco.
· Enzimas
que no cortan: muestra solo
el listado de enzimas de la base de datos que no cortan dentro de la secuencia de DNA problema.
Como
primera medida se procede a ingresar la secuencia de DNA de interés en la caja para tal fin, y acto seguido se hace clic en
CREATE MAP (Figura 48).
Se
puede observar que, en el cuadro de abajo, se muestran todas las enzimas y los sitios donde cortaron la secuencia, si tienen
sitios de restricción que reconocer. Haciendo clic en la enzima indica la secuencia que reconoce (en la barra debajo de la
leyenda ALL RESTRICTION ENZYMES (Figura 48). Al dar clic en el sitio de corte se mostrará el tamaño del fragmento
(Figura 48). Si no se desea ver como gráfico esta información, se hace clic en TEXT DISPLAY (desplegar como texto) (Figura 48). Las enzimas están codificadas de acuerdo a su función, como se indica en
la página, en la sección de información adicional.
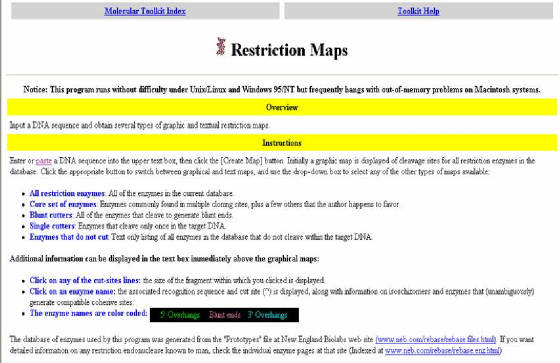
Figura 47. Pagina
del programa Mapas de Restricción. El programa sirve para realizar mapas de restricción de secuencias de DNA, aquí
se presenta las características y forma de utilización.
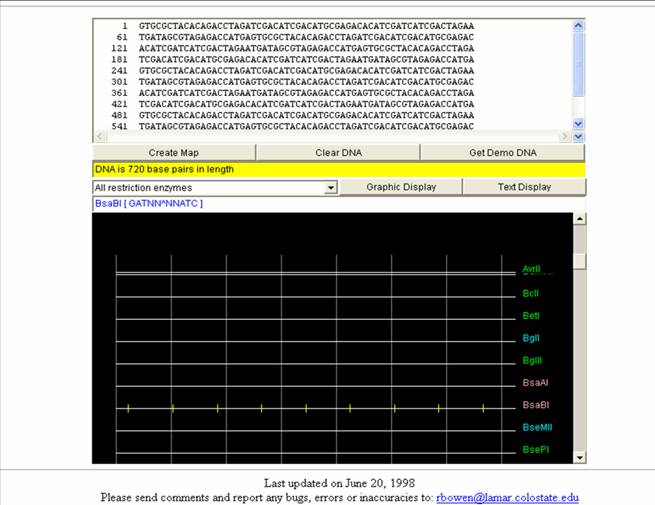
Figura 48. Pagina del programa Mapas de Restricción.
Se observa la manera de ingresar la secuencia de DNA y en la caja negra se encuentran los resultados.
ELECTROFEROGRAMAS-CHROMAS(http://www.technelysium.com.au/chromas.html): Cuando se realizan
ensayos de secuenciación en equipos automatizados, los archivos se pueden abrir con una gran variedad de programas, dependiendo
del sistema operativo del computador. Uno de los más utilizados para Windows es Chromas (Figura 49). Este software despliega
los archivos de cromatógrama que arrojan secuenciadores automatizados de Applied Biosystems y Amersham MegaBace, y archivos
en formato Staden SCF. El programa permite ver la secuencia en ambos sentidos, ampliar o comprimir el gráfico, aplicar colores
a cada base de acuerdo a las necesidades del investigador, observar la secuencia complementaria, traduce la secuencia en 3
ORF, y permite su impresión entre otras ventajas. El programa no es interactivo y requiere bajarlo desde la página, e instalarlo
en el computador.
Figura 49. Pagina del programa Chromas.
COMPARACIÓN DE SECUENCIAS
Los alineamientos múltiples de secuencias son
una herramienta importante en el estudio de las mismas. La información básica que provee es la identificación de regiones
conservadas, lo cual es muy útil en el diseño de experimentos, tales como la evaluación de la función de una proteína, su
modificación o la identificación de nuevos miembros de una familia de genes.
Las secuencias pueden alinearse a través de
toda su longitud (alineamiento global), o solo en ciertas regiones. Los alineamientos globales necesitan usar gaps (que representan
inserciones o delecciones), mientras que los alineamientos locales pueden evitarlos, alineando regiones entre gaps.
Un filograma es un diagrama de ramas (árbol)
donde se asume que es un estimado de una filogenia, las longitudes de las ramas son proporcionales a la cantidad de cambio
evolutivo. Un cladograma es un diagrama con brazos (árbol) donde se asume que es un estimado de la filogenia, los brazos son
de igual longitud; el cladograma muestra el ancestro común, pero no indica la cantidad de tiempo evolutivo que separa las
taxas.
1. Alineamiento múltiple de secuencia-ClustalW (http://www.ebi.ac.uk/clustalw/): ClustalW es un programa interactivo
(también existe la versión para instalar en computador) para alineamiento múltiple de secuencias de DNA o proteínas. Este
genera alineamientos de múltiples secuencias divergentes biológicamente significativas. ClustalW calcula el mejor apareamiento
para las secuencias seleccionadas, y las alinea de tal manera que las identidades, similaridades y diferencias pueden ser
vistas. El alineamiento es progresivo y considera la redundancia de secuencia. Este programa tiene algunos parámetros predeterminados
y ajustables. Las relaciones evolutivas pueden observarse por medio de cladogramas o filogramas.
Cuando
se ingresa al sitio (Figura 50) se abre una ventana que presenta el formulario del programa. Los parámetros preestablecidos
sirven para la mayoría de alineamientos. Tiene una primera casilla (YOUR E-MAIL) donde se coloca el correo
electrónico si se desea que los resultados lleguen por esta vía. La segunda casilla (ALIGNMENT TITLE) permite
colocar un titulo al trabajo. La tercera casilla (RESULTS) nos da la opción de escoger la manera en que se
envíen los resultados (interactivo o por correo electrónico). El siguiente ítem nos posibilita
escoger si el alineamiento se hace completo (FULL) o rápido (FAST). En la opción
KTUP se puede escoger que “longitud de palabra” usar cuando se realizan alineamientos rápidos.
En WINDOW se elige la longitud de la ventana (para alineamientos rápidos). SCORE ayuda a
decidir el puntaje a tener en cuenta en alineamientos rápidos. La casilla TOPDIAG selecciona las diagonales
a integrarse en un alineamiento rápido. En cuanto a PAIGAP se escoge el nivel de gap para alineamientos rápidos. La sección MATRIX sirve para elegir la serie de matrix para el alineamiento (BLOSUM, PAM, GONNET).
La casilla 12 (GAPOPEN) se define el nivel de un gap abierto, los niveles
predeterminados son 15.0 para DNA y 10.0 para proteína. ENDGAP ayuda a escoger el nivel para el cerramiento
de un gap, el nivel predeterminado es –1. GAP EXTENSIÓN determina el nivel de extensión de un gap,
el nivel predeterminado en este ítem para DNA es de 6.66 y 0.2 para proteína. En tanto que GAP DISTANCES
permite elegir el nivel de separación de un gap, el valor predeterminado para esta opción es 4.
OUTPUT decide el formato para el alineamiento (ALN, GCG, PHYLIP, PIR y GDE).
La barra para OUTORDER deja elegir el orden de las secuencias que serán impresas en el alineamiento. Estos
2 ítems regulan también el cálculo del árbol filogenético.
Finalmente,
abajo se encuentra la caja para pegar las secuencias a alinear. También se pueden subir desde el computador con el botón EXAMINAR.
Las secuencias deben estar en el siguiente formato:
>sequence
1
ATGAAGGATGAGGAGAAGATGGAGATTCAGGAGATGCAGCTCAAAGAGGCCAAGCACATT
>sequence
2
GCAGACGACGCANAGGATCGCGCGCAAGGCCTGCAGCGCGAACTGGATGGCGAGCTCTAG
Una vez completado este proceso se hace
clic en RUN (Figura 50).
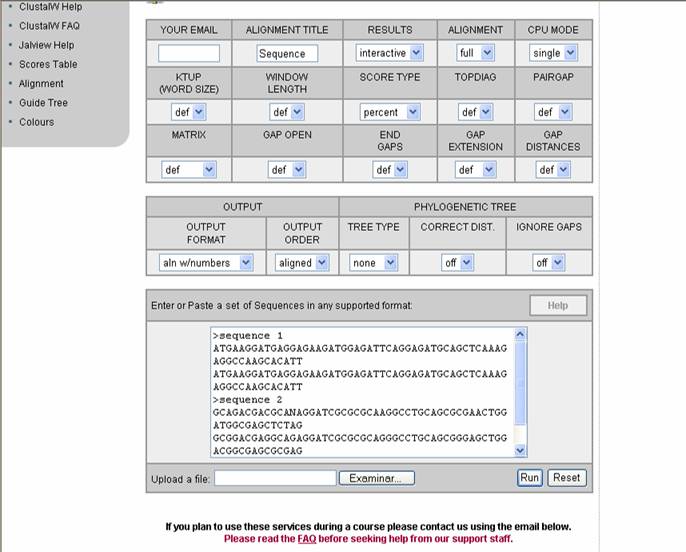
Figura 50. Pagina del programa ClustalW.
Se observa el formato para ingresar las secuencias de DNA.
Luego de lo anterior se abrirá una ventana que mostrará el progreso del
análisis, lo que dará paso a otra ventana con los resultados (Figura 51). El alineamiento puede elegirse con colores, y tiene
unos símbolos que se interpretan así:
· "*" significa que los residuos o nucleótidos en una columna son idénticos en todas
las secuencias del alineamiento.
·
":" significa
que existen sustituciones conservadas de acuerdo al color.
·
"." significa
que sustituciones semiconservadas se han observado.
Los
colores se interpretan de la siguiente forma (para aminoácidos):
AVFPMILW ROJO Pequeño (pequeño hidrofobico (incl. aromático -Y))
DE AZUL Ácido
RHK
MAGENTA Básico
STYHCNGQ VERDE
Hidroxil
+ Amino + Básico - Q
Otros
GRIS
El cladograma se presenta enseguida (Figura
52) y tiene la opción para elegir el filograma.
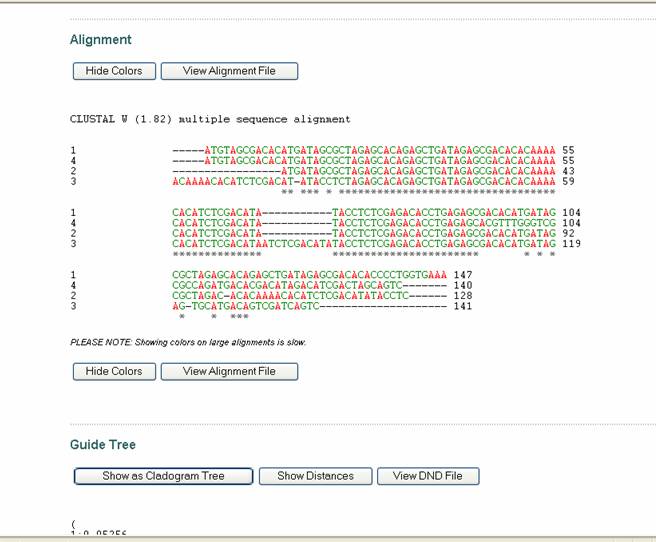
Figura 51. Pagina de resultados del programa ClustalW.
Se observa la presentación en colores de las secuencias de DNA.
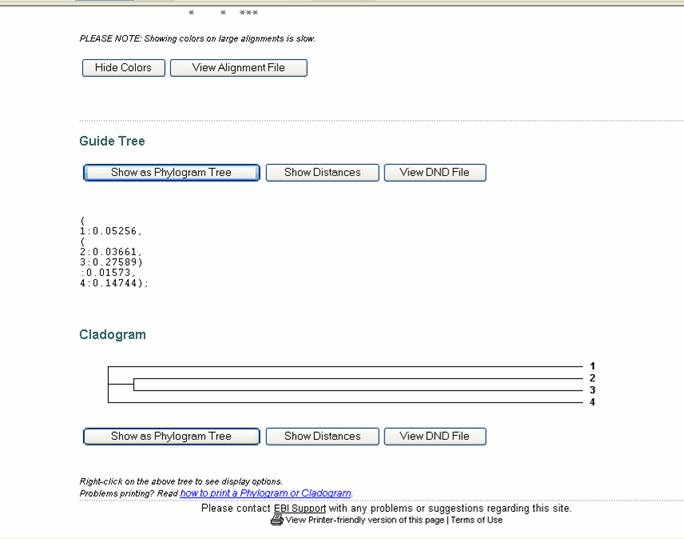
Figura 52. Pagina de resultados del programa ClustalW.
Se observa la presentación del cladograma para las 4 secuencias de DNA ingresadas.
2. Alineamiento múltiple de secuencia-T-Coffee (http://www.ebi.ac.uk/t-coffee/): Este software es otra opción para alineamiento múltiple
de secuencias. T-Coffee es un programa con el
cuál se logran resultados más agudos que con ClustalW, pero tiene la desventaja de que solo maneja un número limitado de secuencias
(menos de 30). Otra característica es que este programa permite combinar resultados obtenidos con otros métodos de alineamiento
(ClustalW, Dialign, etc.), ya que es capaz de combinar esta información para producir un alineamiento acorde con todos los
métodos. El
manejo del programa es similar al que de ClustalW. Básicamente se ingresa las secuencias con el mismo formato ya descrito
para ClustalW, las opciones en la barra superior (E-MAIL, RESULTS, RUN NAME, MATRIZ) se modifican solo si
es necesario de la forma ya descrita para el software ClustalW (Figura 53). Los resultados se presentan de igual manera que
en el programa ClustalW (incluyendo colores, simbología, cladogramas y filogramas).
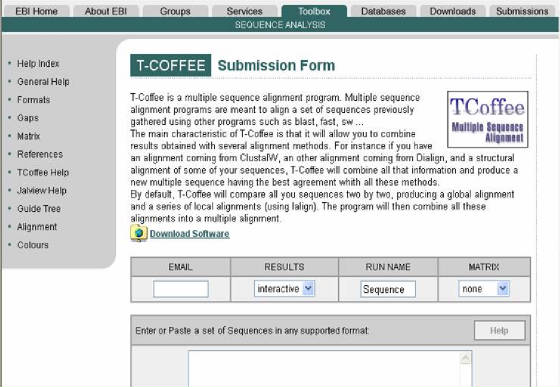
Figura 53. Pagina principal del programa T-Coffee.
DESCARGA DE PROGRAMAS DE BIOINFORMÁTICA
Aunque
existen múltiples programas interactivos, que son aconsejables debido a la actualización permanente de los servidores que
los proveen, hay varios que es necesario descargar desde Internet e instalarlos en el computador. Muchos son de acceso libre,
pero algunos tienen costo (por medio de cargo a tarjeta de crédito).
Aquí
se cita un ejemplo de la manera en que se descarga e instala en la computadora este tipo de programas, ya que la mayoría de
software tienen un procedimiento similar para este fin.
Un
programa muy útil para aplicaciones como pruebas de oligonucleótidos, cortes con enzimas de restricción y traducir la información
de una secuencia de DNA a una secuencia de aminoácidos es FAST PCR. Para su descarga se prosigue así:
Figura 54. Pagina principal del programa Fast PCR
- Luego se da clic en DOWNLOAD (descargar)
-Después, en la siguiente ventana, se
da clic en SELF-EXTRACTED
WINRAR ARCHIVE INSTALLATION FILE HERE: FASTPCR.EXE
2.5MB) (Figura 55).
- A partir de allí se siguen las directrices
que da el computador. Se
escoge la carpeta donde se va a guardar el programa
(preferiblemente archivos de programa o Mis Documentos) (Figura
55).
- Seguidamente, se iniciará la descarga del programa y al final se
generará un vínculo en la barra de programas (Figura 56). Al dar clic
allí se abrirá la ventana principal del programa listo para su utilización
(Figura 57).
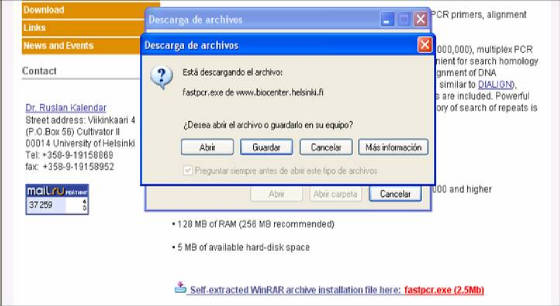
Figura 55. Ventana de instalación del programa Fast PCR.
Se puede observar el link de inicio de instalación “Self-extracted winrar archive
installation file here: fastpcr.exe (2.5mb)” (abajo en azul y rojo), y la ventana auxiliar donde se elige la carpeta
donde se almacenara el programa.
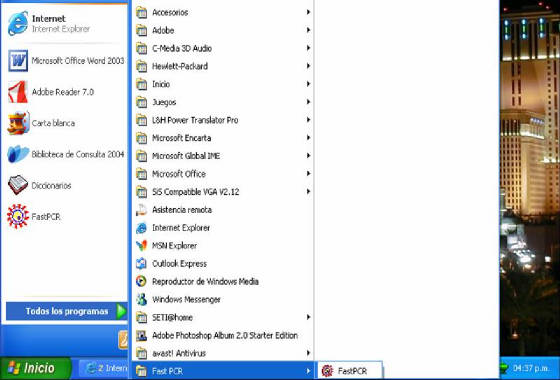
Figura 56. Icono de acceso al programa Fast PCR.
En la parte inferior del menú “Todos los programas” de la sección “inicio” del computador se puede
observar el link de inicio del software FastPCR.
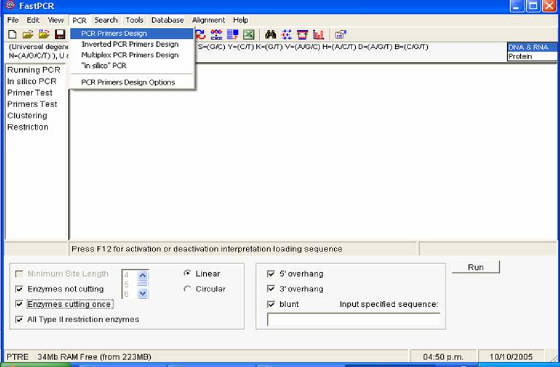
Figura 57. Ventana principal del programa Fast PCR.
Se puede observar el menú desplegado con las funciones que realiza el software.
El
software Fast PCR esta diseñado para utilizar secuencias para aplicaciones de PCR: PCR estándar y PCR extendida, PCR inversa,
PCR degenerada, PCR múltiple, PCR “in silico”, pruebas de primers y calculo de la temperatura de melting optima
para productos de PCR desconocidos. Aquí se describe brevemente el programa (existe un manual en línea al que se accede por
medio del menú HELP).
- El programa trae
una caja donde se pega la secuencia problema de DNA y una barra de herramientas en la parte superior con las funciones del
programa, (Figura 57).
- Si se desea diseñar un oligonucleótido se da clic en la ventana
PCR y luego en PCR PRIMERS DESIGN. Las características de los primers se especifican haciendo clic en el
último logo de la barra de iconos (Figura 57).
- Cuando el objetivo es trasladar una secuencia de DNA, a secuencia
de aminoácidos, se pega la secuencia problema en la caja y se da clic en el logo correspondiente de la barra de iconos (Figura
58).
- Para realizar cortes virtuales con enzimas de restricción se hace clic en RESTRICTION
en la parte izquierda de la pantalla, después de haber pegado la secuencia problema de DNA en la caja. Una vez hecho esto
se procede a marcar todas las opciones de enzimas de restricción en la parte baja de la pantalla (Figura 58).
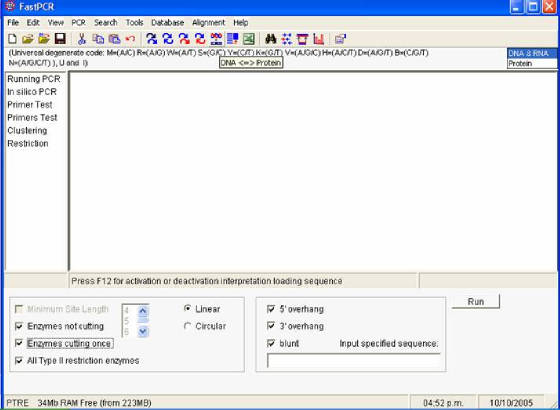
Figura 58. Ventana del programa Fast PCR. Se puede
observar el logo para traducir secuencias de DNA. Abajo están las opciones para las enzimas de restricción.
|